pachydermoperiostosis













Pachydermoperiostosis (PDP) refers to a rare osteoarthrodermopathic syndrome with familial and idiopathic forms. It is also known as primary hypertrophic osteoarthropathy because it is not due to a secondary cause such as lung disease.
Terminology
Also known as Touraine-Solente-Golé syndrome, named after the three French physicians who described it in 1935 (see below).
Epidemiology
PDP most commonly affects black people and adolescent boys.
Clinical presentation
PDP is characterized by marked thickening of the skin of the extremities, face, and scalp (including cutis verticis gyrata).
Pathology
PDP is an autosomal dominant inherited disorder that is self-limiting and progresses for several years by stabilizing.
Radiographic features
Plain radiograph/CT
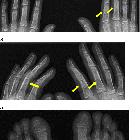
Skeletal findings include symmetric, shaggy subperiosteal bone formation in the long bones, especially of the forearm and leg bones, but also of the metacarpals, metatarsals, and phalanges.
Involvement of the epiphyseal region distinguishes it from the secondary form, in which the epiphyses are usually spared .
There can be a widening of the ends of bones, especially in the wrist and knee joints which are due to increased bone formation. The generalized and symmetric periosteal reaction in pachydermoperiostosis tends to blend with the cortex and primarily involves the distal ends of the radius, ulna, tibia, and fibula.
History and etymology
Although not the first to describe the entity, the French physicians, Albert Touraine (1883-1961), G Solente and L Golé, were the first to recognize pachydermoperiostosis as a primary form of hypertrophic osteoarthropathy in 1935 .
Siehe auch:
und weiter:


 Assoziationen und Differentialdiagnosen zu primäre hypertrophe Osteoarthropathie:
Assoziationen und Differentialdiagnosen zu primäre hypertrophe Osteoarthropathie:
