mucopolysaccharidosis type II
Hunter syndrome, also known as mucopolysaccharidosis type II (MPS II) is a rare genetic mucopolysaccharidosis disorder characterized by specific clinical features .
Epidemiology
Hunter syndrome is an X-linked recessive disease and therefore much more common in males. It is a rare disorder with a prevalence estimated at 1 in 170,000 male births .
Clinical presentation
Clinical features occur across a wide spectrum of severity, ranging from mild to very severe. Features are not present at birth but develop around 24-48 months.
The disorder shares some similarities with Hurler syndrome (MPS I) including:
- abnormal facies: prominent forehead, flattened nasal bridge, coarse facies, macroglossia, gingival hyperplasia
- short stature
- characteristic skeletal abnormalities: dysostosis multiplex, macrocephaly
- cognitive deficits
- cardiovascular disorders including left ventricular hypertrophy, hypertension, and arrhythmia that may progress to congestive cardiac failure
- hepatosplenomegaly
- deafness
- skin lesions, which include distinctive pearly papules over the scapulae and lateral upper arms
Clinical features are similar to Hurler syndrome (MPS I) but are differentiated by lack of corneal clouding .
Pathology
Etiology
Hunter syndrome is an X-linked recessive disorder caused by mutations in the iduronate 2-sulfatase (IDS) gene, responsible for the degradation of heparan and dermatan sulfate. Mutations of IDS lead to accumulation of proteoglycan products in tissues, leading to organ dysfunction and growth abnormalities .
Radiographic features
Hurler syndrome and other mucopolysaccharidoses are associated with characteristic skeletal abnormalities collectively known as dysostosis multiplex.
- thoracolumbar kyphosis
- hip dysplasia: poor acetabular development
- genu valgum deformity
- spinal cord compression if severe
- enlargement of the skull
- thick calvarium
- broad clavicles and ribs
- extremities show hypoplastic epiphyses and thickened coarse diaphyses
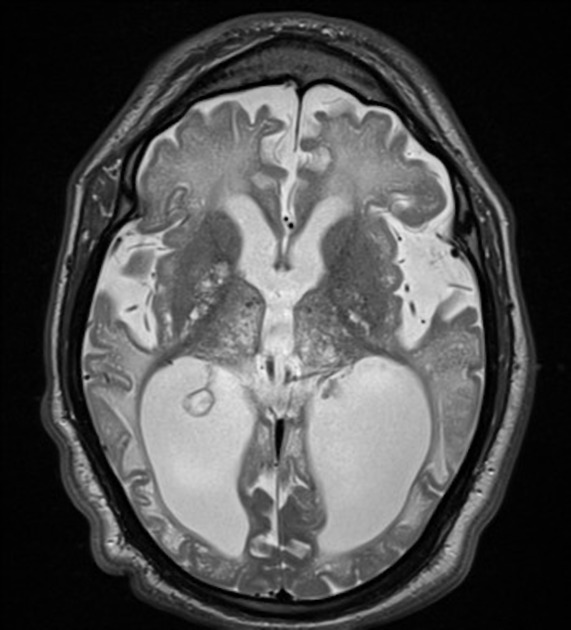
MRI
MRI Brain often displays:
- hydrocephalus and venticulomegaly
- enlarged subarachnoid spaces
- patchy distribution of white and grey matter signal intensity abnormalities, affecting multiple lobes and reducing contrast between grey and white matter on T2-weighted imaging
Treatment and prognosis
The mainstay of treatment for mucopolysaccharidoses is enzyme replacement therapy (ERT). For Hunter syndrome, this consists of replacement with idursulfase administered via infusions. Enzyme replacement therapy in Hunter syndrome has been shown to produce symptomatic improvement in somatic symptoms and appears to slow disease progression . However, because current enzyme replacement therapy is unable to cross the blood-brain barrier it does not affect cognitive decline . Hematopoietic cell transplantation has been shown to improve clinical outcomes but also does not prevent cognitive decline .
Unfortunately despite best medical therapies the disease is progressive in nature. Patients with more severe forms of Hunter syndrome may survive to their teens or early twenties, and those with mild forms may have normal intelligence and survive into their sixth or seventh decade .
History and etymology
Hunter syndrome is named after Charles Hunter, the physician who first described the disease and its characteristic clinical manifestations in 1917 .
Siehe auch:
und weiter:


 Assoziationen und Differentialdiagnosen zu Mukopolysaccharidose Typ II:
Assoziationen und Differentialdiagnosen zu Mukopolysaccharidose Typ II: