Wolman disease
Wolman disease is a rare autosomal recessive inborn error of metabolism resulting in the deposition of fats in multiple organs.
Clinical presentation
Patients with Wolman disease typically present during the first two months of life with failure to thrive, diarrhea and vomiting. Abdominal distention may be present because of hepatomegaly, splenomegaly and generalized lymphadenopathy .
Pathology
The underlying biochemical abnormality is a deficiency of acid lipase/acid esterase. These enzymes are responsible for splitting triglycerides and cholesterol esters . The deficiency results in accumulation of lipid esters in multiple tissues including the liver, spleen, lymph nodes, and small bowel.
Marked accumulation of cholesterol and fatty acid crystals also occurs within the cells of the adrenal cortex, resulting in enlargement of the adrenal glands. Saponification (accumulation of glyceryl ether lipids) of this tissue with subsequent calcification results in the characteristic radiological features.
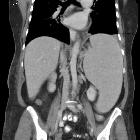
Radiographic features
Plain radiograph
- bilateral calcification of the adrenal glands, which are enlarged
CT
- may show hepatosplenomegaly (with fatty liver)
- bilaterally enlarged calcified (punctate calcification) adrenal glands that retain their normal triangular shapes
- may show enlarged fatty-infiltrated lymph nodes
MRI
- hepatosplenomegaly
- bilateral adrenal enlargement
- bilateral adrenal calcifications
- T1: low signal foci
- T2: low signal foci
Treatment and prognosis
Treatment approaches have included total parenteral nutrition (TPN), corticosteroids, plasma infusion, and various dietary supplements, all without success, with death in infancy ensuing (usually within 6 months). Bone marrow transplantation has been reported with some success.
History and etymology
It takes its name from Moshe Wolman (1914-2009) , an Israeli neuropathologist, who first described the entity in 1956.
Differential diagnosis
For adrenal calcification see: differential diagnosis for adrenal gland calcification
Siehe auch:
und weiter:


 Assoziationen und Differentialdiagnosen zu Wolman disease:
Assoziationen und Differentialdiagnosen zu Wolman disease: