Pelizaeus-Merzbacher disease

Pelizaeus-Merzbacher disease (PMD) is an X-linked leukodystrophy which is characterized by an arrest in myelin development.
Clinical presentation
Patients may present with
- pendular eye movements (nystagmus)
- hypotonia
- pyramidal disease
- ataxia
Pathology
Genetics
Pelizaeus-Merzbacher disease is the result of abnormalities of the proteolipidprotein (PLP1) gene locus at Xq22. This can be either a mutation, deletion or duplication (most common) .
Subtypes
Traditionally Pelizaeus-Merzbacher disease has divided into two subtypes:
- classic
- connatal: more rare and severe
Radiographic features
CT
CT features, as is the case with many leukodystrophies, are non-specific and may show hypoattenuating white matter with progressive white matter atrophy.
MRI
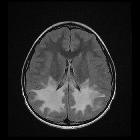
Features include:
- T1: lack of myelination, often seen as low T1 signal regions typically involving internal capsule, proximal corona radiata and the optic radiation
- T2: near complete absence of expected low signal in the supratentorial region
- abnormal signal can either be diffuse or patchy
- if there is patchy involvement, a characteristic tigroid appearance may be seen
- may also show cortical sulcal prominence due to atrophy
- white matter volume may be decreased
- may also involve cerebellum and brainstem
- MR spectroscopy
- affected areas often show a reduction in the NAA peak
- decreased choline (due to hypomyelination)
History and etymology
Named after Friedrich Christoph Pelizaeus (1851 - 1942), a German neurologist, and Ludwig Merzbacher (1875–1942), an Italian neuropathologist and psychiatrist.
Differential diagnosis
Although numerous other leukodystrophies are in the general differentials, specific entities to be considered include:
- Pelizaeus-Merzbacher-like disease (mutations of GJA12 at 1q41-q42 or MCT8 at Xq13.2)
- 18q- deletion (18q22.3 q23)
- Salla disease (SLC17A5 gene at 6q14-q15).
Siehe auch:
und weiter:


 Assoziationen und Differentialdiagnosen zu Pelizaeus Merzbacher disease:
Assoziationen und Differentialdiagnosen zu Pelizaeus Merzbacher disease: