Pseudolaminar necrosis





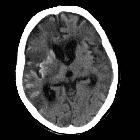


Cortical laminar necrosis, also known as pseudolaminar necrosis, is necrosis of neurons in the cortex of the brain in situations when the supply of oxygen and glucose is inadequate to meet regional demands. This is often encountered in cardiac arrest, global hypoxia and hypoglycemia.
Terminology
Cortical laminar necrosis and pseudolaminar necrosis, although often used interchangeably, have distinct meanings histologically :
- cortical laminar necrosis refers to involvement of the entire cortex
- pseudolaminar necrosis refers to selective involvement of mid and deep layers (3, 4 and 5)
Unfortunately, both terms are often used inappropriately for a broader range of ischemic events which result in areas of cortical T1 intrinsic hyperintensity, or cortical enhancement, or eventual cortical dystrophic calcification.
Although the underlying reason for changes at a cellular level are presumably the same (i.e. liquefactive necrosis involving cortex with influx of monocytes and phagocytosis of cellular debris), it is the overall pattern of cellular damage (i.e. restricted to the cortex) that distinguishes cortical laminar and pseudolaminar necrosis from other more regional forms of ischemic damage (e.g. thromboembolic cerebral infarction) .
Clinical presentation
Cortical laminar or pseudolaminar necrosis tend to be seen in patient who have had events that impair the ability to provide the brain with sufficient nutrients for its required function. This can be due to either reduced supply (e.g. hypoxia, ischemia, and sometimes hypoglycemia) or increased demand (e.g. status epilepticus) .
More specifically it is encountered in:
- cerebral hypoperfusion
- generalized secondary to cardiac arrest or hypotension
- watershed (borderzone) distribution due to hypotension and stenosis of relevant vessel (typically internal carotid artery)
- hypoxia
- hypoglycemia
- hematological diseases such as severe anemia
- status epilepticus (as the brain demands more glucose and oxygen)
- cyanide poisoning
Pathology
Cortical laminar necrosis occurs as a consequence of the neurons within the cortex being far more metabolically active than glial cells or adjacent white matter. Additionally, not the whole cortex is similarly at risk. There is selective vulnerability of certain layers of the cerebral neocortex (cortical layers 3, 4, and 5) to metabolic stress, as well as certain regions of the brain (e.g. primary visual cortex and perirolandic cortex).
The selective vulnerability of grey matter may be due to higher metabolic demand and denser concentration of receptors for excitatory amino acids that are released after an anoxic-ischemic event.
Radiographic features
Laminar necrosis may be identified within hours of the anoxic-ischemic event. In the acute phase DWI is superior to conventional MRI sequences to distinguish these cortical changes .
CT
Appearances of cortical laminar necrosis on CT can be subtle, appearing as gyriform changes in attenuation, both hypodense and hyperdense depending on timing. No hemorrhage or calcification is evident acutely. After a few days, gyral enhancement will be seen which typically persists for up to 3 months.
MRI
Although early cytotoxic edema causes high signal seen on DWI with corresponding low apparent diffusion coefficient (ADC) values in the affected cortex, and cortical enhancement may be seen later, typically after 2 weeks, intrinsic T1 signal increase is the most specific imaging feature.
T1 curvilinear hyperintensities signaling laminar necrosis become evident as early as 3 to 5 days after stroke, but typically after 2 weeks, with a peak of intensity around one month, and then slowly fades usually over 3 or so months . Occasionally it can remain visible for over a year after the insult . This T1 high signal is believed to be caused by the accumulation of denatured proteins in dying cells and/or lipid laden macrophages. Importantly it does not represent the presence of hemorrhage or calcium .
T2 weighted images demonstrate either increased signal or iso-intensity to unaffected cortex .
See also
Siehe auch:
und weiter:


 Assoziationen und Differentialdiagnosen zu kortikale laminäre Nekrose:
Assoziationen und Differentialdiagnosen zu kortikale laminäre Nekrose: