idiopathic interstitial pneumonias

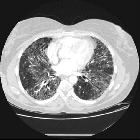

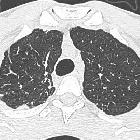
The idiopathic interstitial pneumonias (IIPs) are diffuse interstitial lung diseases of unknown cause. They are characterized by cellular infiltration of the interstitial compartment of the lung with varying degrees of inflammation and fibrosis.
Classification
Over the years many attempts have been made to reach a consensus on classification of the idiopathic interstitial pneumonias (IIPs). This imposed a great deal of confusion and difficulty not only for clinicians and radiologists but also medical students. The most recent widely accepted classification was developed by the American Thoracic Society (ATS) and the European Respiratory Society (ERS) in 2002 and revised in 2013. The initial classification was mainly based on the histologic findings and divided IIPs into seven subtypes:
- usual interstitial pneumonia (UIP)/idiopathic pulmonary fibrosis (IPF)
- idiopathic non-specific interstitial pneumonia (NSIP)
- cryptogenic organizing pneumonia (COP): formerly bronchiolitis obliterans organizing pneumonia (BOOP)
- respiratory bronchiolitis–associated interstitial lung disease (RB-ILD)
- desquamative interstitial pneumonia (DIP)
- lymphoid interstitial pneumonia (LIP)
- acute interstitial pneumonia (AIP): the only acute process in the list
In the revised version, a multidisciplinary approach with integration of all information provided by HRCT chest, clinical findings and course of the disease, as well as histopathologic correlation is used to reach the final diagnosis. Even after exhausting all inputs, if clear distinction cannot be made (e.g. even on biopsy, histologic features of more than one subtype may be present) the diagnosis is then considered as "unclassifiable" IIP.
In the 2013 ATS/ERS revision, IIPs are assigned into four major groups:
- chronic fibrosing IIPs: IPF and idiopathic non-specific interstitial pneumonia
- smoking related IIPs: respiratory bronchiolitis–associated interstitial lung disease and desquamative interstitial pneumonia
- acute and subacute IIPs: acute interstitial pneumonia and cryptogenic organizing pneumonia
- rare IIPs: idiopathic pleuroparenchymal fibroelastosis (added in 2013 revision) and lymphoid interstitial pneumonia
Radiographic features
Optimal imaging protocol for assessment of patients with suspected IIP is crucial and should be adjusted in each case. Traditional non-contiguous HRCT chest is widely replaced by volumetric image acquisition mainly due to better assessment of the distribution, apicobasal gradient, patchiness of the disease, presence of bronchiectasis, or pulmonary nodules. Ideal acquisition time is at the end of full inspiration when total lung capacity is reached.
Multi-detector HRCT chest with thin collimation and sharp preprocessing algorithm (bone algorithm) and thin (less than 2 mm) multiplanar reconstruction is desired. Prone images are helpful to exclude dependent atelectasis obscuring or mistaken for underlying peripheral fibrotic changes.
Expiratory images, usually in non-contiguous acquisition, is crucial to assess air trapping which usually implies alternative diagnosis such as hypersensitivity pneumonitis rather than IIP.
Siehe auch:
- Kryptogene organisierende Pneumonie (COP)
- lymphozytisch interstitielle Pneumonie
- gewöhnliche interstitielle Pneumonie (UIP)
- Interstitielle Lungenerkrankung
- non specific interstitial pneumonia (NSIP)
- Akute interstitielle Pneumonie
- pleuroparenchymale Fibroelastose
- What Every Radiologist Should Know about Idiopathic Interstitial Pneumonias
- Desquamative interstitielle Pneumonie (DIP)
- respiratory bronchiolitis–associated interstitial lung disease
- fibrotic idiopathic interstitial pneumonia
- All Idiopathic Chronic Lung Diseases aRe Nonspecific
und weiter:


 Assoziationen und Differentialdiagnosen zu Idiopathische interstitielle Pneumonie:
Assoziationen und Differentialdiagnosen zu Idiopathische interstitielle Pneumonie: