lymphocytic interstitial pneumonitis

















Lymphocytic interstitial pneumonitis is a benign lymphoproliferative disorder characterized by lymphocyte predominant infiltration of the lungs. It is classified as a subtype of interstitial lung disease. It also falls under the umbrella of non-lymphomatous pulmonary lymphoid disorders.
Epidemiology
Lymphocytic interstitial pneumonitis can occur at any age. However, most of the patients are adults with a mean age of 50 years. If a child presents with lymphocytic interstitial pneumonitis, this can be indicative of AIDS.
There is a recognized female predilection most likely attributable to the fact that lymphocytic interstitial pneumonitis occurs in patients with autoimmune disease such as Sjögren syndrome, which is by far more common in women .
Clinical presentation
The main clinical symptoms are a gradual onset of dyspnea and cough with approximately six months duration. Less frequently, patients may have systemic symptoms such as fever, night sweat, arthralgia, and weight loss. If the disease progresses to the end-stage respiratory failure cyanosis and clubbing may develop. Hypertrophy of the salivary glands observed in 20% of patients .
Pathology
There is diffuse infiltration of the interstitium and alveolar spaces by lymphocytes and plasma cells.
Associations
- Sjögren syndrome: can occur in up to 25% of those with lymphocytic interstitial pneumonitis
- AIDS: particularly if it occurs in the young
- autoimmune thyroid disease
- systemic lupus erythematosus
- Castleman disease
- common variable immune deficiency
- rheumatoid arthritis
- pulmonary amyloidosis
Markers
In about 80% of patients polyclonal or IgM monoclonal gammopathy is found .
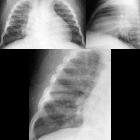
Radiographic features
Plain radiograph
Features can be non-specific, but may include:
- lower-zone predominant bilateral reticular opacification
- chronic bilateral airspace opacification
CT
The following features may be seen with lymphocytic interstitial pneumonitis on HRCT, but the findings are not exclusive to its diagnosis:
- features tend to be diffuse with mid to lower lobe predominance
- thickening of bronchovascular bundles
- interstitial thickening along lymph channels
- small but variable sized pulmonary nodules (can be centrilobular or subpleural, and are often ill-defined)
- ground-glass changes
- scattered thin-walled cysts
- usually deep within the lung parenchyma
- typically abut vessels (i.e. is perivascular or subpleural)
- size range between 1-30 mm (useful for differentiation from lymphoma of the lung )
- mediastinal lymphadenopathy
Treatment and prognosis
The natural history is variable, from near-complete resolution to progressive disease. More than 30% of patients will develop the end-stage disease and honeycombing despite treatment.
Transformation to lymphoma can occur, particularly in a patient with monoclonal gammopathy or hypogammaglobulinemia . Corticosteroids have been successfully trialled .
Differential diagnosis
General imaging differential considerations include:
- pneumocystis pneumonia (PCP)
- cystic changes (pneumatoceles) seen in advanced disease
- can be difficult to differentiate particularly in those with AIDS
- lymphangioleiomyomatosis
- occur in younger females
- cysts are generally uniformly distributed throughout the lungs
- Langerhans cell histiocytosis
- smokers
- bizarre cysts that spare the costophrenic angles
- upper lung zone predominant
See also
Siehe auch:
- Rheumatoide Arthritis
- Hypersensitivitätspneumonitis
- systemischer Lupus Erythematodes
- Morbus Castleman
- Sjögren-Syndrom
- Pneumocystis jiroveci Pneumonie
- Hashimoto-Thyreoiditis
- Pneumonitis
- Variables Immundefektsyndrom
- AIDS
und weiter:
- Lymphangiosis carcinomatosa
- Bronchiektasen
- Milchglasverschattungen
- verdickte interlobuläre Septen
- multiple zystische Lungenherde
- idiopathic interstitial pneumonia (mnemonic)
- thickening of bronchovascular bundles
- Interstitielle Lungenerkrankung
- interstitial pneumonia
- differential of chronic alveolar opacities
- retikuläres Muster
- AIDS defining illness
- perilymphatische Lungennoduli
- chronic bilateral airspace opacification
- What Every Radiologist Should Know about Idiopathic Interstitial Pneumonias
- pulmonary manifestations of AIDS
- Idiopathische interstitielle Pneumonie
- non lymphomatous pulmonary lymphoid disorders
- lymphoproliferative Lungenerkrankungen
- Lymphoproliferative Erkrankung nach Transplantation


 Assoziationen und Differentialdiagnosen zu lymphozytisch interstitielle Pneumonie:
Assoziationen und Differentialdiagnosen zu lymphozytisch interstitielle Pneumonie: