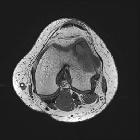
langerhans cell histiocytosis























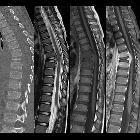
Langerhans cell histiocytosis (LCH) is a rare multisystem disease with a wide and heterogeneous clinical spectrum and variable extent of involvement.
Terminology
Langerhans cell histiocytosis was previously known as histiocytosis X. The newer term is preferred as it is more descriptive of its cellular background, and removes the ambiguity of the connotation "X".
Epidemiology
The disease is more common in the pediatric population, with a peak incidence between one and three years of age . Incidence is estimated at ~5 per million children, and 1-2 cases per million adults . There is also a male predilection (M:F ~ 1.5:1) .
Clinical presentation
Essentially any part of the body can be affected and as such, clinical presentation will depend on specific involvement. The course of the disease ranges from those that spontaneously regress to those that have a rapidly progressive course (the latter is especially common in young children with multisystem disease).
Historically, three (two eponymous) forms have been recognized, although there is some confusion as to their definition :
- Letterer-Siwe disease
- disseminated multiorgan disease
- typically young children/infants less than one year old
- fulminant course with poor prognosis
- Hand-Schüller-Christian disease
- multiple lesions
- some authors confine the term to patients with solitary organ involvement
- other authors accept multi-organ involvement (e.g. bone and spleen)
- confined to the one location (usually bone)
- typically children
- intermediate prognosis
- multiple lesions
- eosinophilic granuloma (EG)
- lesions are confined to one organ system
- some authors confine the term to patients with a solitary lesion
- other authors accept multiple lesions
- 70% of cases affect bone
- typically children
- best prognosis
- lesions are confined to one organ system
A more useful and less controversial classification, which roughly correlates to the eponymous diseases above, is as follows:
- multiple organ systems, multiple sites involved
- single organ system, multiple sites involved
- single lesion
Additionally, in 2008 the WHO recommended distinguishing LCH from a more pleomorphic variant known as Langerhans cell sarcoma .
As well as systemic disease, individual organ systems may be involved, which will be discussed separately:
- skeletal manifestations of LCH
- central nervous system manifestations of LCH
- hepatobiliary manifestations of LCH
- pulmonary manifestations of LCH
- salivary gland manifestations of LCH
- gastrointestinal manifestations of LCH
The remainder of this article concerns a general overview of LCH.
Pathology
Langerhans cell histiocytosis is due to uncontrolled monoclonal proliferation of Langerhans cells (distinctive cells of monocyte-macrophage lineage) and should be considered a malignancy although its biological behavior is very variable . An immune-mediated mechanism has been postulated. This proliferation is accompanied by inflammation and granuloma formation. Electron microscopy may reveal characteristic Birbeck granules. Immunohistochemistry reveals expression of the following antigens:
- HLA-DR
- CD1a
- CD207 (langerin)
- S100
Radiographic features
Imaging features are often not pathognomonic and tissue diagnosis is usually required for definitive diagnosis. As langerhans cell histiocytosis can affect most organ systems, radiographic appearances are discussed separately (see above).
Treatment and prognosis
The prognosis can be extremely variable with eosinophilic granuloma carrying the best and Letterer-Siwe disease carrying the worst prognosis. The prognosis is more closely related to the disease burden rather than histological features, although frankly malignant features (Langerhans cell sarcoma) do also have an impact on survival :
- unifocal disease (eosinophilic granuloma): >95% survival
- two organ involvement: 75% survival
- Langerhans cell sarcoma: 50% survival
History and etymology
The Langerhans cell was discovered within the epidermis by German physician Paul Langerhans (1847-1888) in 1865 when he was a medical student and working under famed Professor Rudolf Virchow .
See also
- Erdheim-Chester Disease: a non-Langerhans cell histiocytosis
- cystic angiomatosis
- Rosai-Dorfman disease: sinus histiocytosis
- juvenile xanthogranuloma
- hemophagocytic lymphohistiocytosis
- 2016 Histiocyte Society classification of histiocytoses
Siehe auch:
- Langerhanszell-Histiozytose der Lunge
- Erdheim-Chester-Erkrankung
- skeletale Manifestationen der Langerhanszell-Histiozytose
- Rosai-Dorfman-Erkrankung
- zystische Angiomatose
- Hand-Schüller-Christian-Syndrom
- Langerhans' cell histiocytosis of the liver
- Langerhans-Zell-Histiozytose der Wirbelsäule
- pulmonary manifestations of LCH
und weiter:
- Osteolysen der Kalotte
- Läsionen der Felsenbeinspitze
- Cholesteatom
- verdickter Hypophysenstiel
- diffuses intrinsisches Ponsgliom
- erworbenes Cholesteatom
- Cholesteatom des äußeren Gehörgangs
- Gibbus
- miliare Lungenherde
- Flüssigkeit in den Mastodzellen
- causes of pulmonary arterial hypertension
- radiologisches muskuloskelettales Curriculum
- single layer periosteal reaction
- Knochentumoren
- conditions involving skin and bone
- PLCH
- Histiozytose
- Hand-Schuller-Christian disease
- pulmonary arterial hypertension classification - third world symposium on PAH
- mass involving the foramen of Monro or/and superior third ventricle
- upper lobe pulmonary fibrosis (mnemonic)
- langerhans cell
- intracerebral Langerhans cell histiocytosis
- Langerhans-Zell-Histiozytose der Orbita
- langerhans cell histiocytosis with osseous and pulmonary involvement
- solitary eosinophilic granuloma of the radius
- Histiozytosis X der Leber
- langerhans cell histiocytosis of the thyroid gland
- Metastasen in der Hypophyse


 Assoziationen und Differentialdiagnosen zu Histiozytose X:
Assoziationen und Differentialdiagnosen zu Histiozytose X: