Erdheim-Chester-Erkrankung








































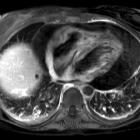
Erdheim-Chester disease (ECD) is a rare non-Langerhans cell, non-familial multisystemic histiocytosis, with widespread manifestations and of highly variable severity. The most common presenting symptom is bone pain.
Epidemiology
Erdheim-Chester disease is a rare, non-inherited disease of middle age with a slight male predominance .
Clinical presentation
Patients may present with a variety of symptoms, ranging from focal neurological deficits to multiorgan failure . The most common presenting symptom is bone pain. Patients may also present with focal neurological signs, exophthalmos, retroperitoneal fibrosis, diabetes insipidus, and dyspnea due to extraskeletal involvement of these systems.
Pathology
Erdheim-Chester disease is a systemic lipogranulomatous disorder with infiltration by lipid-laden histiocytes (foamy macrophages), Touton giant cells and a variable amount of background fibrosis . In contrast to Langerhans cell histiocytosis (LCH), no S-100 nor CD1 are detected , but CD68 is positive .
Both Erdheim-Chester disease and LCH may coexist, and cases of double infiltration have been reported .
Radiographic features
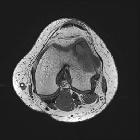
Musculoskeletal involvement is most common, with multifocal extraskeletal involvement seen in 30-50% of patients .
Skeletal involvement
- bilateral, symmetric metaphyseal and diaphyseal sclerosis
- increased uptake on Tc-MDP bone scan
- cortical thickening
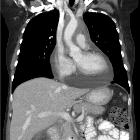
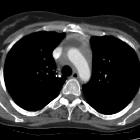
Visceral
- lung (see: pulmonary manifestations of Erdheim-Chester disease)
- can appear similar to Langerhans cell histiocytosis with predominantly cystic disease, septal thickening and preserved lung volume
- chest radiographs will often show interstitial edema pattern (interlobular septal thickening) with cardiomegaly and pleural effusions that do not respond to diuretics
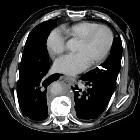
- kidneys and retroperitoneum
- often involved
- usually asymptomatic
- hairy kidney sign: irregular symmetric infiltration of the bilateral perirenal and posterior pararenal spaces
- coated aorta sign: periaortic soft tissue
- inferior vena cava and pelvic ureters are typically spared, which are useful cross-sectional imaging findings for differentiation of retroperitoneal Erdheim-Chester disease from retroperitoneal fibrosis
- skin
- retro-orbital tissue
- optic nerve edema
- retrobulbar masses that can cause proptosis and motility impairment
- retrograde extension along the optic nerve to the hypothalamus may explain the distribution of brain involvement
- heart, pericardium and aorta
Intracranial
Intracranial involvement of the dura, brain and pituitary are rare :
- meninges
- dural accumulations may mimic meningiomas, with enhancing soft tissue masses
- T2 signal characteristics are somewhat different, as the accumulations in Erdheim-Chester disease are hypointense
- brain: usually affecting the hypothalamus ; intraparenchymal masses in ECD appear non-specific
- pituitary infundibulum: presenting with diabetes insipidus
Treatment and prognosis
Steroids, radiotherapy and chemotherapy have all been used but with little effect, with some patients relentlessly progressing . Pulmonary fibrosis and cardiac failure are the most common causes of death . Given the small volume of published data, mortality rates are sketchy but may be as high as 60% .
Surgical or percutaneous intervention for hydronephrosis, orbital or meningeal involvement is useful for symptomatic local disease.
History and etymology
It was first described in 1930 as "lipid granulomatosis" by Jakob Erdheim (1874-1937), an Austrian pathologist, and William Chester (1903–1974), an American pathologist .
Differential diagnosis
The differential diagnosis for Erdheim-Chester disease is very dependent on location although some entities will be considerations in most locations (e.g. lymphoma).
Intracranial
The differential for intracranial involvement is that of other causes of dural masses including:
Perinephric
See also
Siehe auch:
- Meningeom
- verdickte interlobuläre Septen
- Histiozytose X
- Rosai-Dorfman-Erkrankung
- Histiozytose
- akute monozytische Leukämie
- Niemann-Pick-Krankheit
- Erdheim-Chester-Erkrankung Manifestation an der Niere
- diffuse perirenale Raumforderungen
- juvenile xanthogranuloma
- Hashimoto-Pritzker-Syndrom
- Seeblaue Histiozytose
- Maligne Histiozytose
- Hämophagozytische Lymphohistiozytose
und weiter:
- Osteopoikilose
- Tumoren der Hypophysenregion
- Neurosarkoidose
- Lymphom der Niere
- Tuberkulose des ZNS
- Pituitary MRI (an approach)
- dysthyroid eye disease
- tram-track sign
- hypertrophe Pachymeningitis
- Tuberkulom des ZNS
- verdickter Nervus Opticus
- tuberkulöse Pachymeningitis
- ZNS-Beteiligung bei Erdheim-Chester-Erkrankung
- Zeichen der ummantelten Aorta


 Assoziationen und Differentialdiagnosen zu Erdheim-Chester-Erkrankung:
Assoziationen und Differentialdiagnosen zu Erdheim-Chester-Erkrankung: