Kearns-Sayre syndrome


Kearns-Sayre syndrome (KSS), also known as oculocraniosomatic disorder, is a rare multisystem mitochondrial disorder.
Clinical presentation
The patient often presents with progressive external ophthalmoplegia . Neurologic symptoms develop in childhood or adolescence, usually before 20 years of age .
Other features include:
- cardiac conduction defects
- retinal pigmentation
- ptosis
Pathology
The disease is characterized by the ragged-red appearance of muscle fibers, and the presence of mitochondrial DNA (mtDNA) with large deletions in affected tissues. It tends to affect peripheral white matter early and preferential involvement of the globi pallidi and thalami.
Genetics
It is sporadic in a majority of cases.
Radiographic features
CT
CT scan of the brain shows basal ganglia siderocalcific deposits and subcortical calcifications (with or without basal ganglia deposits), and diffuse supratentorial and infratentorial atrophy.
MRI
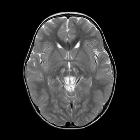
Cerebral, cerebellar and brainstem atrophy are mainstay features of the disease .
Signal characteristics include:
- T2: subcortical prolongation with subcortical calcifications, with or without bilateral basal ganglia siderocalcific deposits.
History and etymology
It was initially described by Thomas P Kearns and George Pomeroy Sayre in 1958 .
Differential diagnosis
Imaging differential considerations include
- other mitochondrial disorders, e.g. Leigh syndrome
- chronic progressive external ophthalmoplegia (CPEO): some consider KSS as a syndromic variant of CPEO
Siehe auch:
und weiter:


 Assoziationen und Differentialdiagnosen zu Kearns-Sayre-Syndrom:
Assoziationen und Differentialdiagnosen zu Kearns-Sayre-Syndrom: