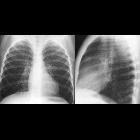
neuroblastoma




















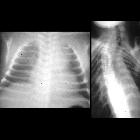


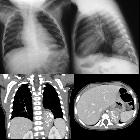
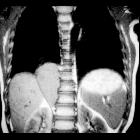



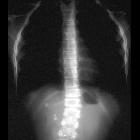

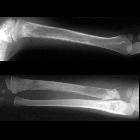







Neuroblastomas are tumors of neuroblastic origin. Although they may occur anywhere along the sympathetic chain, the vast majority arise from the adrenal gland.
They represent the most common extracranial solid childhood malignancy and are the third commonest childhood tumor after leukemia and brain malignancies. They account for ~15% of childhood cancer deaths.
Epidemiology
The tumors typically occur in infants and very young children (mean age of presentation being ~22 months) with 95% of cases diagnosed before the age of 10 years. Occasionally, they may be identified antenatally or immediately at birth (see congenital neuroblastoma) .
Clinical presentation
Typically with pain or a palpable mass and abdominal distension, although numerous other presentations may be encountered due to local mass effect.
Other accompanying syndromes include:
- Hutchinson syndrome: skeletal metastases may present with skeletal pain or limping and irritability or proptosis with periorbital and cranial bumps
- Pepper syndrome: hepatomegaly due to extensive liver metastasis
- blueberry muffin syndrome: multiple cutaneous lesions
- opsomyoclonus : rapid, involuntary conjugate fast eye movements
- proptosis and periorbital ecchymoses ("raccoon eyes"): orbital metastases
Pathology
The tumors arise from the primitive neuroectodermal cells or neural crest cells (adrenal medulla precursor). The histology is similar to small round blue cell tumors . The majority of them demonstrate chromosome 1p deletion and N-myc amplification.
Macroscopically, they tend to be large grey-tan colored soft lesions, with or without fibrous pseudocapsule; hence, some are well defined, and some are infiltrative. Areas of necrosis, hemorrhage, and particularly calcification, are very common.
Microscopically, they form Homer Wright rosettes . Most of them secrete catecholamines: vanillylmandelic acid (VMA) and homovanillic acid (HVA) .
Location
Neuroblastomas arise from the sympathetic nervous system :
Intra-abdominal disease (two-thirds of cases) is more prevalent than intrathoracic disease. Specific sites include:
- adrenal glands: most common site of origin, 35%
- retroperitoneum: 30-35%
- organ of Zuckerkandl
- celiac axis
- paravertebral sympathetic chain
- posterior mediastinum: 20%
- neck: 1-5%
- pelvis: 2-3%
Associations
The vast majority of neuroblastomas are sporadic; however, in rare instances, they may be associated with :
- Beckwith-Wiedemann syndrome
- central failure of ventilation
- DiGeorge syndrome
- Hirschsprung disease
- neurofibromatosis type 1
Radiographic features
Plain radiograph
Appearances are non-specific, typically demonstrating an intrathoracic soft-tissue mass or an intra-abdominal mass displacing adjacent organs. Pressure on adjacent bones may cause remodeling of ribs, vertebral bodies or pedicle thinning. Up to 30% may have evidence of calcification on the plain film.
Skeletal metastases are usually ill-defined and lucent, with periosteal reaction or metaphyseal lucency. Sclerotic metastases are uncommon .
Ultrasound
Neuroblastoma on ultrasound demonstrates a heterogeneous mass with internal vascularity. Often there are areas of necrosis that appear as regions of low echogenicity. Calcification may or may not be evident on ultrasound .
CT
On CT, the tumor typically is heterogeneous with calcifications seen in 80-90% of cases . Areas of necrosis are of low attenuation.
The tumor morphology is often helpful, with the mass seen insinuating itself beneath the aorta and lifting it off the vertebral column. It tends to encase vessels and may lead to compression. Adjacent organs are usually displaced, although in more aggressive tumors direct invasion of the psoas muscle or kidney can be seen. The latter can make distinguishing neuroblastoma from Wilms tumor difficult (see neuroblastoma vs. Wilms tumor).
Lymph node enlargement is often present.
MRI
MRI is superior to all other modalities in assessing the organ of origin, intracranial or intraspinal disease and bone marrow disease .
- T1: heterogeneous and iso to hypointense
- T2
- heterogeneous and hyperintense
- cystic/necrotic areas very high intensity
- signal voids may be evident
- C+ (Gd): variable and heterogeneous enhancement
Nuclear medicine
Some compounds are used for diagnosis and staging:
- pentetreotide labeled to Indium-In (a somatostatin analog)
- not specific for neuroblastic tissue
- MIBG (metaiodobenzylguanidine labeled to Iodine-)
- 95% of neuroblastomas secrete catecholamines, however, 10-30% of neuroblastomas are negative on MIBG
- sensitivity: 88%
- specificity: 99% (for sympathetic tissue)
- does not distinguish between neuroblastoma, ganglioneuroblastoma, ganglioneuroma, carcinoid, and pheochromocytoma
- FDG PET/CT
Surveillance for metastatic recurrence:
- Tc-m MDP
- 36% of primary tumors negative
- mainly to evaluate skeletal metastases
- also able to detect some lung and liver metastases
Staging and metastatic disease
For staging refer to neuroblastoma staging.
Metastatic disease is common and has a variety of patterns:
- bone
- most common
- liver
- diffuse infiltration (more common in stage 4S)
- focal hypo-enhancing masses
- lung and pleura
- discrete nodules
- diffuse consolidation
- pleural disease is uncommon
- brain and meninges
- dural metastases can be diffuse or nodular
- brain metastases are uncommon but variable in appearance
Treatment and prognosis
Treatment depends on the patient's stage. Localized tumors considered to be 'low-risk' are surgically excised, and patients tend to do very well (see below). In 'high-risk' tumors, a combination of surgery, chemotherapy +/- bone marrow transplantation is employed, unfortunately with poor overall results. In some cases, where tumors are very large, pre-surgical chemotherapy to attempt to downstage the tumor may be administered .
Patients with stage 1, 2, or 4S have a better prognosis. Unfortunately 40-60% of patients present with stage 3 or 4 diseases . For advanced disease, the age of the child is most important .
- stage 1, 2 or 4S: 75-90% 3 year survival
- stage 3
- <1 year of age: 80-90% 1-year event-free survival
- >1 year of age: 50% 3-year survival
- stage 4
- <1 year of age: 60-75% 1-year event-free survival
- >1 year of age: 15% 3-year survival
Poor prognostic factors
- later age of onset: >18 months
- higher stage: particularly in the presence of metastasis
- N-Myc mutation
- chromosome 1p deletion
- unfavorable Shimada histology index
Better prognostic factors
- TRK-A expression
Differential diagnosis
For intrathoracic neuroblastoma consider:
- intrathoracic lymphoma
- extralobar pulmonary sequestration
- round pneumonia
- ganglioneuroma
- ganglioneuroblastoma
For intra-abdominal neuroblastoma consider:


 Assoziationen und Differentialdiagnosen zu Neuroblastom:
Assoziationen und Differentialdiagnosen zu Neuroblastom: