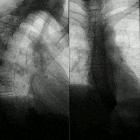
Fibrodysplasia ossificans progressiva




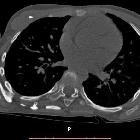











Fibrodysplasia ossificans progressiva (FOP), previously known as myositis ossificans progressiva (MOP) and also known as Munchmeyer’s disease, is a rare, inherited disorder characterized by progressive fibrosis and ossification of muscles, tendons, fasciae, aponeuroses, and ligaments of multiple sites. It is disabling and ultimately fatal.
FOP should not be confused with myositis ossificans circumscripta, which is usually posttraumatic.
Epidemiology
FOP is a very rare disease, with an estimated prevalence of ~1 per 2 million. There is no racial/ethnic, or gender predilection . The process and symptoms of heterotopic ossification start between 2 and 5 years old.
Pathology
Most cases arise from sporadic mutations, although some are inherited in an autosomal dominant fashion .
Radiographic features
FOP can be evaluated using plain radiographs, CT and/or MRI. MRI is mostly useful for more subtle edema which would not be seen on the other modalities. Characteristic features include:
- congenital hallux valgus
- monophalangic first toe
- shortened metacarpals
- pseudoexostoses (ossification of ligamentous insertions)
- microdactyly of the first metacarpal/metatarsal
- neck muscle edema
- C2-C7 facet joint fusion
Treatment and prognosis
This is a progressive, fatal disease with the median survival being 45 years.
Differential diagnosis
Since the process of heterotopic ossification is quite unique, differential diagnosis on a long term basis is quite limited. However, for punctual abnormalities, the following differential diagnosis should be considered:
- scleroderma
- CREST syndrome
- juvenile fibromatosis
- dermatomyositis
Siehe auch:
- Myositis ossificans
- Dermatomyositis
- Systemische Sklerodermie
- Heterotope Ossifikation
- CREST syndrome
- Calcinosis cutis universalis
und weiter:


 Assoziationen und Differentialdiagnosen zu Fibrodysplasia ossificans progressiva:
Assoziationen und Differentialdiagnosen zu Fibrodysplasia ossificans progressiva: