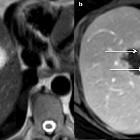
polycystic liver disease

Polycystic liver disease (PCLD) is a hereditary condition that may arise either in patients with autosomal dominant polycystic kidney disease (ADPKD) or in patients with a different genetic mutation that results solely in autosomal dominant polycystic liver disease.
Clinical presentation
Most patients are asymptomatic but occasionally patients can have pain with large cysts. Other uncommon symptoms include abdominal distention, early satiety and orthopnea.
Pathology
It is characterized by progressive development of fluid-filled biliary epithelial cysts throughout all segments of the liver.
75-90% of patients with autosomal dominant polycystic kidney disease have PCLD. PCLD without ADPKD has a prevalence of <0.01% .
Treatment and prognosis
The course of polycystic liver disease is variable but progressive. In patients with autosomal dominant polycystic kidney disease, the number and size of cysts increases with advancing age .
Treatment for symptomatic cysts is usually based on the morphology of the liver cysts. Fenestration is considered a safe and acceptable procedure for patients with a dominant cyst pattern where liver size can be reduced after the cysts collapse. A combination of resection-fenestration is suitable for those with a diffuse cyst pattern where grossly affected segments are resected in combination with fenestration to allow for reduction in liver size .
Siehe auch:
- autosomal-dominante polyzystische Nierenerkrankung
- Differenzialdiagnosen zystischer Leberläsionen
- zystische Lebermetastasen
- simple Leberzyste
- posttraumatic hepatic cyst
und weiter:


 Assoziationen und Differentialdiagnosen zu Zystenleber:
Assoziationen und Differentialdiagnosen zu Zystenleber: