autosomal-dominante polyzystische Nierenerkrankung










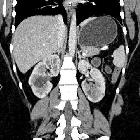
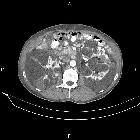



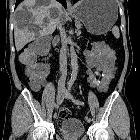
















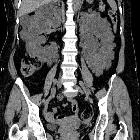

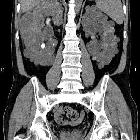

Autosomal dominant polycystic kidney disease (ADPKD), also sometimes more vaguely referred to as "adult polycystic kidney disease", is as the name would suggest, a hereditary form of adult cystic renal disease.
Epidemiology
Autosomal dominant polycystic kidney disease is one of the most common serious hereditary diseases, found in 1:400 to 1:1000 individuals, and by far the most common hereditary cause of end stage renal failure (ESRF) . It accounts for 4-10% of all cases of ESRF .
Clinical presentation
The kidneys are normal at birth, and with time develop multiple cysts. At the age of 30 years, approximately 68% of patients will have visible cysts by ultrasound . That figure increases over time, such that essentially all patients eventually demonstrate cystic change. By the age of 60 years, approximately 50% of patients have end stage renal failure. The risk of renal cancer is not increased.
Clinical presentation is variable and includes :
- dull flank pain of variable severity and time course: most common
- abdominal or flank masses
- hematuria
- hypertension: usually develops at the same time as renal failure
- renal functional impairment to renal failure
Pathology
Macroscopically the kidney demonstrates a large number of cysts of variable size (from a few mm to many cms), in both the cortex and medulla. They are filled with fluid of variable color (from clear or straw colored to altered blood or chocolate colored to purulent when infected).
Associations
A number of conditions are well recognized as being associated with ADPKD :
- cerebral berry aneurysms
- found in 6% of patients with ADPKD without a family history of aneurysms
- found in up to 16% of patients with ADPKD with a family history
- intracranial dolichoectasia: 2-3%
- hypertension: up to 80% adults
- colonic diverticulosis
- small bowel diverticula (perhaps)
- bicuspid aortic valve
- mitral valve prolapse: up to 25%
- aortic dissection
- multiple biliary hamartomas (von Meyenberg complexes)
- cysts in other organs
- liver: most common, 75% by age 60 years
- ovaries
- spleen: ~5 %
- seminal vesicles: 60% by age 40 years
- prostate: 11%
- pancreas: ~10%: N.B. pancreatic cysts are more common in von Hippel Lindau disease (vHL)
Genetics
The majority of cases are inherited in an autosomal dominant fashion. In a minority of cases, no family history is present, and the disease is due to a spontaneous mutation .
Two genes have been identified, with slightly different phenotypes :
- PKD1
- located on chromosome 16p
- 85% of cases
- presentation is earlier and more likely to progress to end stage renal failure (ESRF)
- PKD2
- located on chromosome 4q
- 15% of cases
- less severe
A third rare type of ADPKD (termed ADPKD 3) has been described, however, the gene has yet to be identified .
The defect results in cystic dilatation of the renal tubules (of all parts of the nephron) in a minority of nephrons. The cysts are variable in size and result in compression of the remainder of the kidney, resulting in increased renin and erythropoietin secretion, and gradual renal dysfunction.
Radiographic features
Imaging of patients with autosomal dominant polycystic kidney disease can be challenging, simply due to the size and number of the cysts and associated mass effect on adjacent structures. It is potentially tedious, but necessary, to assess all cysts for atypical features, that may reflect complications (e.g. hemorrhage or infection) or malignancy (i.e. renal cell carcinoma) .
Plain radiograph
Plain films have no role in the surveillance of patients with established ADPKD. The diagnosis may be suspected when the renal outlines are enlarged, multilobulated or difficult to discern, with associated displacement of loops of bowel.
Multiple calcifications may be seen which may have multiple ring configurations.
On intravenous urography, "Swiss cheese nephrogram" is seen due to multiple radiolucencies noted as a result of multiple renal cysts. "Spiderleg pyelogram" is also described since stretched out and attenuated pelvicalyceal system is seen as a result of mass effect caused by renal cysts.
Ultrasound
Ultrasound is an excellent choice for repeated imaging as it is fast, relatively inexpensive and lacks ionizing radiation. It is able both to suggest the diagnosis and to assess for cyst complications.
Simple renal cysts will appear anechoic with well-defined imperceptible walls, posterior acoustic enhancement (amplification) and lateral shadowing (extinction) .
Cysts with hemorrhage or infection will demonstrate echogenic material within the cyst, without internal blood flow. Calcification may develop. Renal cell carcinomas in contrast, although usually cystic in the setting of ADPKD, will have solid components of thick septa with blood flow.
Perinephric hematomas may be visible and collections of variable echogenicity surrounding the kidney.
Additionally, ultrasound is also able to visualize cysts in other abdominal organs.
CT
CT is of course very sensitive to the diagnosis and excellent at characterizing renal cysts. Simple cysts appear as rounded structures with near water attenuation (~ 0 HU). The wall is very thin and regular, and are often imperceptible.
Cysts which have had internal complications may be hyperattenuating, with internal non-enhancing septations and/or calcifications.
A complex cystic mass with solid components or thick septa which enhance should be viewed with suspicion, and presence of a renal cell carcinoma (RCC) suspected (see Bosniak classification of renal cysts).
MRI
Renal cysts appear morphologically the same as on CT, rounded well-defined structures with very thin regular walls .
- T1: low signal
- T2: high signal
- T1 C+ (Gd)
- although MRI has exquisite contrast resolution, care must be taken prior to administering gadolinium-based contrast agents due to the risk of nephrogenic systemic fibrosis
- simple cysts should not have any solid enhancing components
- presence of enhancement of a solid component or septa should raise the possibility of a renal cell carcinoma (RCC) (N.B. infected cysts may peripherally enhance, as do islands of trapped renal tissue)
Treatment and prognosis
ESRF requiring transplant or dialysis eventually develops in many patients (45% by the age of 60). Patients with PKD1 mutations are more likely to progress to ESRF and often do so at an earlier age .
Complications
Complications may be both local (i.e. of the kidney) or of other organ systems.
Renal complications include :
- progression to end stage renal failure
- recurrent urinary tract infections
- cyst hemorrhage or infection-resulting in acute pain
- cyst rupture: resulting in retroperitoneal hemorrhage
Unlike in some other congenital cystic kidney diseases, there is no increased risk for renal cell carcinoma (RCC) unless the patient is undergoing prolonged dialysis .
Distant complications include:
Differential diagnosis
General imaging differential considerations include:
- von Hippel Lindau disease (vHL)
- pancreatic cysts more common
- primary renal disease related cysts
- the renal parenchyma appears abnormal, reduced in volume with increased echogenicity on ultrasound
- multiple 'incidental' renal cysts
- usually far fewer in number
- kidney not enlarged
- hydronephrosis mimicking multiple cysts
- acquired cystic kidney disease: occurs in those with chronic renal failure (particularly in those on dialysis)
- autosomal recessive polycystic kidney disease (ARPKD)
- enlarged kidney
- cysts are very numerous and small
- changes are present in childhood
- corticomedullary differentiation is lost
- medullary cystic disease
- cysts smaller and located in the medulla cortex junction
- multicystic dysplastic kidney
- may be unilateral, unlike ADPKD
Practical points
- it is often useful for the nephrologist if the radiologist provides the total kidney volume (TKV) for both kidneys
- one of the most common TKV calculators in use (the Mayo calculator) relies on four different measurements: an AP measurement, a TRV measurement, a long axis measurement in the sagittal plane, and a long axis measurement in the coronal plane
See also


 Assoziationen und Differentialdiagnosen zu autosomal-dominante polyzystische Nierenerkrankung:
Assoziationen und Differentialdiagnosen zu autosomal-dominante polyzystische Nierenerkrankung:





