fabry disease









Fabry disease, also known as Anderson-Fabry disease, is a multisystem disorder resulting from an X-linked inborn error of metabolism and is a lysosomal storage disorder. The disease results from genetic mutations that cause decreased or absent expression of hydrolase alpha-galactosidase A, ultimately resulting in abnormal accumulation of globotriaosylceramide (Gb3) in various organ systems .
The clinical manifestations are variable and may depend on the specific gene defect and degree of residual enzyme activity. Both male and heterozygous females can exhibit severe phenotype.
Clinical presentation
Fabry disease was initially described in males with a form of severe disease, a phenotype known as a "classic" Fabry. However, it is now recognized that there are both early and late-onset forms of the disease in males, depending on the genetic aberration and degree of enzymatic compromise .
Similarly, females that have heterozygous genetic involvement have a spectrum of presentation ranging from asymptomatic to severe. This may be explained by a variable expression of the mutated X-linked gene .
Neurological
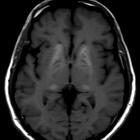
Small vessel ischemia is the main mechanism of central nervous system manifestations, with ischemic strokes, especially of the posterior circulation being common. Multi-infarct dementia can ensue, although MRI T2 hyperintensities in the white matter of the frontal and parietal lobes can be seen in asymptomatic patients. MRI is used to monitor treatment response.
T1 weighted images also demonstrate high signals in the deep grey matter especially that of the pulvinar, relating to mineralization (see basal ganglia T1 hyperintensity). Exclusive involvement of pulvinar is thought to be characteristic of the condition .
Additionally, basilar artery dolichoectasia is characteristic of this disease .
Both T2 and T1 changes have been seen to regress with treatment if instituted early enough.
Acroparesthesia is a common manifestation, more so than ischemic changes described above, and can be debilitating .
Autonomic (sympathetic) nervous system involvement can lead to gastrointestinal autonomic dysfunction.
Renal
Renal involvement begins with proteinuria progressing to end-stage renal failure usually in the 4 decade. On imaging, the kidneys have non-specific findings of medical renal disease including increased echogenicity, thinned renal cortex, and multiple renal cysts. The cysts are perhaps the most specific sign, typically small and of uniform size, located just beneath the capsule, aiding in differentiating these from autosomal dominant polycystic kidney disease (ADPKD).
Ocular
Fabry disease is affiliated with corneal verticillata and lenticular abnormalities. Recent studies have proposed eye signs in Fabry disease in association with α-galactosidase A mutations could be an indicator of disease severity .
Cardiac
Cardiac involvement is frequent within the scope of the ‘classic phenotype’ and is common in isolation. It is the leading cause of death in Fabry disease.
See cardiac manifestations of Fabry disease
Musculoskeletal
Avascular necrosis of the femoral head has been described.
Pulmonary
There can be chronic obstructive airways disease like symptoms with bronchial wall thickening.
Treatment and prognosis
Enzyme substitution (hydroxylase alpha-galactosidase) is efficacious in rectifying the metabolic deficit. It consists of an intravenous infusion which is typically given every two weeks . Enzyme replacement has been shown to improve some deposition-related components of the disease, including some reversal in cardiac enlargement and clearance of excessive glycolipids from renal podocytes .
A modestly-sized (n=110) retrospective Dutch study showed the median life expectancy of 57 years for males and 72 for females .
History and etymology
First described in 1898 by Johannes Fabry, German dermatologist (1860-1930) .
Siehe auch:
- Aseptische Knochennekrose
- Ischämischer Schlaganfall
- basal ganglia T1 hyperintensity
- autosomal-dominante polyzystische Nierenerkrankung
- Chronisch obstruktive Lungenerkrankung
- acroparesthesia
- pulvinar sign
und weiter:


 Assoziationen und Differentialdiagnosen zu Morbus Fabry:
Assoziationen und Differentialdiagnosen zu Morbus Fabry: