interrupted aortic arch
















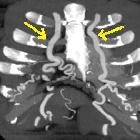




Interrupted aortic arch (IAA) is an uncommon congenital cardiovascular anomaly where there is a separation between the ascending and descending aorta. It can either be complete or connected by a remnant fibrous band. An accompanying large ventricular septal defect (VSD) and/or patent ductus arteriosus (PDA) is frequently present.
Epidemiology
It may account for ~1.5% of congenital cardiac anomalies.
Pathology
Faulty embryological development of the aortic arch (thought to occur during the 5 to 7 week of intrauterine life).
Classification
According to the Celoria-Patton classification, IAA can be classified into three types according to the location of the anomaly:
- type A: second most common, the interruption occurs distal to the left subclavian arterial origin
- type B: most common (>50%), the break occurs between the left common carotid and left subclavian arterial origins
- type C: rare, interruption occurs proximal to the left common carotid arterial origin
Each type is divided into three subtypes :
- subtype 1: normal subclavian artery
- subtype 2: aberrant subclavian artery
- subtype 3: isolated subclavian artery that arises from the ductus arteriosus
Associations
- DiGeorge syndrome
- found commonly in those with a type B interruption
- almost always associated if there is a right-sided descending aorta
- truncus arteriosus
- aortopulmonary septal defect (aortopulmonary window)
- transposition of the great arteries
- double outlet right ventricle
- functional single ventricle
Radiographic features
Plain radiograph
Plain film features are often non-specific:
- the aortic knuckle may be absent
- may show cardiomegaly
Antenatal ultrasound/Echocardiography
The right ventricle may appear a lot larger than the left, although this is a non-specific finding. The ascending aorta may also appear more vertical than usual. These modalities may not allow differentiation of IAA from severe aortic coarctation with a hypoplastic arch .
CT
Allows visualization of the interrupted aortic arch and associated anomalies.
MRI
- non-visualization of the portion of interruption
- great vessels may show a "V" configuration on coronal imaging
Treatment and prognosis
If uncorrected, it carries a very poor prognosis with extrauterine survival being as little as a few days. Prostaglandin E1 may be given to initial management to keep the ductus open. Surgical correction (either single- or multistage) is the definitive treatment.
Differential diagnosis
General differential considerations include:
- short segment severe aortic coarctation
Siehe auch:
und weiter:


 Assoziationen und Differentialdiagnosen zu Atresie der Aorta:
Assoziationen und Differentialdiagnosen zu Atresie der Aorta: