Mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes (MELAS)











Mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes (MELAS) is one of many mitochondrial disorders. As mitochondria, which have their own DNA, are exclusively passed on from the mother these disorders are only inherited from the mother.
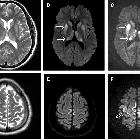
On imaging, it manifests as multifocal stroke-like cortical lesions in different stages of evolution ("shifting spread" pattern), crossing the cerebral vascular territories, and showing a certain predilection to the posterior parietal and occipital lobes. MR spectroscopy may demonstrate elevated lactate in an otherwise normal appearing brain .
Epidemiology
As the long name suggests, MELAS is characterized by 'stroke-like' episodes, typically in childhood or early adulthood (90% present before 40 years of age).
Clinical presentation
MELAS usually has a relapsing-remitting course, with or without superimposed accretion of permanent deficits. Clinical presentation is characterized by :
- most common
- stroke-like episodes
- seizures
- lactic acidosis
- encephalopathy
- dementia
- muscle weakness
- deafness
Pathology
The defect involves the respiratory chain (responsible for energy production). A point mutation at nucleotide 3243 mtDNA (A to G translocation) which encodes for transfer RNA (tRNA) for leucine is the most common cause of the condition. It is therefore thought that this abnormality results in abnormal protein production throughout the mitochondria and affects multiple parts of the respiratory chain. The exact mechanism notwithstanding, the net result is depletion of NAD+ and NADH+. This, in turn, results in a shift to anaerobic metabolism accounting for the buildup of lactic acid and renders the cortex susceptible to neuronal death .
As some mitochondria are passed in the ovum, not all will have the mutant mtDNA. The percentage of mutated genes will affect the severity of clinical manifestations .
Diagnosis
To make the diagnosis of MELAS identification of the most common pathogenic mtDNA variant (m.3243A>G) can be made on peripheral blood samples in 80% of patients. To identify non-m.3243A>G mutations additional testing or muscle biopsy may be required .
Radiographic features
CT
- multiple infarcts
- involving multiple vascular territories
- may be either symmetrical or asymmetrical
- parieto-occipital and parieto-temporal involvement is most common
- basal ganglia calcification
- more prominent feature in older patients
- atrophy
MRI
- acute infarcts
- chronic infarcts
- involving multiple vascular territories
- may be either symmetrical or asymmetrical
- parieto-occipital and parieto-temporal most common
MR spectroscopy: may demonstrate elevated lactate in otherwise normal appearing brain parenchyma or in CSF .
Digital subtraction angiography (DSA)
- usually normal
- enhancing gyri, presumably due to the breakdown of the blood-brain barrier and reperfusion hyperemia correlating with a blush on angiography
Differential diagnosis
Possible differential considerations include:
- other mitochondrial disorders
- status epiliepticus
- viral encephalitis
- cerebral vasculitis
- Creutzfeldt-Jakob disease
- infarcts, due to
Siehe auch:
- Leigh-Syndrom
- CADASIL
- Moyamoya-Erkrankung
- posteriores reversibles Enzephalopathiesyndrom
- Creutzfeldt-Jakob-Krankheit
- Mitochondropathie
- Kearns-Sayre-Syndrom
- status epiliepticus
und weiter:


 Assoziationen und Differentialdiagnosen zu Mitochondriale Encephalomyopathie mit Lactatacidose und Schlaganfall-ähnlichen Episoden (MELAS):
Assoziationen und Differentialdiagnosen zu Mitochondriale Encephalomyopathie mit Lactatacidose und Schlaganfall-ähnlichen Episoden (MELAS):