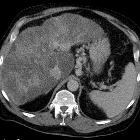
primary hepatic lymphoma


Primary hepatic lymphoma (PHL) is rare, with roughly 100 described cases. If it is being considered as a diagnosis, distal lymphadenopathy, splenomegaly, bone marrow disease, and leukemia should not be present for at least 6 months after the liver tumor is detected (see: secondary hepatic lymphoma) .
Epidemiology
Rare presentation of lymphoma (<1% of all Non-Hodgkin lymphomas).
In contrast, according to one source, diffuse large B-cell lymphoma (DLBCL) affects around 7 out of 100,000 people annually in the United States, with potential for secondary involvement of the liver, so this is a much more common cause of hepatic lymphoma.
Clinical presentation
The most common presentation includes abdominal pain, jaundice, and weight loss. Although the majority of patients will have elevated liver enzymes, these may be normal. The typical age of presentation is around the age of 55. It has a male preponderance.
Pathology
The exact cause of this primary hepatic lymphoma is still unclear, however, it is primarily found in immunocompromised patients such as AIDS/HIV, congenital immunodeficiency, immunosuppressant therapy for organ transplant and collagen vascular disease. Reports have also shown infections as a cause such as EBV, HIV, hepatitis C and hepatitis B with chronic hepatitis and cirrhosis.
The majority of cases of primary liver lymphoma are diffuse large B-cell lymphoma. Other histologic types include peripheral T-cell leukemia/lymphoma-not-otherwise-specified, follicular lymphoma, mantle cell lymphoma, or Burkitt lymphoma.
Radiographic features
Primary hepatic lymphoma is most commonly seen as a solitary mass (53-65%). PHL does not have characteristic radiographic features and can often be mistaken for other liver tumors. Liver biopsy is the most specific way of making the diagnosis.
Ultrasound
Ultrasound is usually the initial radiological test performed:
- typically hypoechoic to the liver parenchyma, probably due to high cellularity and lack of background stroma
- rarely, they may be so hypoechoic that they may appear similar to a cyst
- occasionally have a "target" appearance with a central hyperechogenicity and outer hypoechogenicity
- increased peripheral vascularity with color Doppler
CT
- frequently a single (~60%), solid, hypoattenuating (due to hypovascularity) lesion as opposed to secondary lymphoma which can be more diffusely infiltrating
- there can be a large variation in size, but PHL tends to have dominant masses, as opposed to secondary lymphomatous involvement of the liver, which tends to be more diffuse
- variable contrast enhancement on CT, but tend to be hypoenhancing relative to liver on all phases, with no enhancement in some cases
- an ill-defined mass at the porta hepatis is possible
- hemorrhage, necrosis, and rim-enhancement are possible
MRI
MRI findings depend on the extent of infiltration.
- T1: typically hypointense relative to remainder of liver
- T2: often shows variable intensity
- T1 C+ (Gd): there is poor gadolinium enhancement of these lesions, but they may show faint peripheral rim enhancement
- DWI/ADC: restricts diffusion
Signal characteristics are almost identical to secondary lymphomatous involvement but manifestation as a single solid mass may favor towards a primary hepatic lymphoma .
Nuclear medicine
PET/CT
PET/CT is useful in making the diagnosis and can also be used for staging.
- FDG-PET
- avid FDG uptake of the lymphomatous mass in the liver
- can show or exclude extrahepatic sites of lymphoma with a high degree of sensitivity
Treatment and prognosis
PHL is primarily treated with chemotherapy. The most common chemotherapy regimen for advanced diffuse large B-cell lymphoma (DLBCL) is called R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone). In the United States, this regimen is generally given every three weeks for 6 to 8 cycles. Other treatment options include radiation therapy or surgery.
Differential diagnosis
- secondary hepatic involvement of the liver from Non-Hodgkin lymphoma: much more common
- infection/abscess in a patient receiving chemotherapy
- hepatocellular carcinoma: more common liver mass
- metastatic disease to the liver: patient without known lymphoma
Siehe auch:
- Leberzirrhose
- Lymphom
- Lymphom der Leber
- Lebertumoren
- secondary hepatic lymphoma
- HIV-related primary hepatic lymphoma
- AIDS
und weiter:


 Assoziationen und Differentialdiagnosen zu primary hepatic lymphoma:
Assoziationen und Differentialdiagnosen zu primary hepatic lymphoma: