pulmonary alveolar proteinosis



























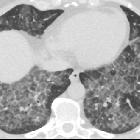
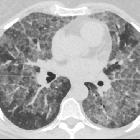


Pulmonary alveolar proteinosis (PAP) is a lung disease characterized by an abnormal intra-alveolar accumulation of surfactant-derived lipoproteinaceous material.
On imaging, PAP is classically associated with the lung crazy paving pattern on CT, although it is a rare cause of this non-specific finding.
Epidemiology
Pulmonary alveolar proteinosis is rare and usually presents in young and middle-aged adults (20-50 years of age) . Smoking is strongly associated with the condition, and in smokers, there is a recognized male predilection (M:F of ~2:1) , which is absent in non-smoking patients .
When the disease presents before the age of 1 year, there is an association with thymic alymphoplasia .
Clinical presentation
Clinical presentation is usually with non-specific respiratory symptoms such as dyspnea or a minimally-productive cough. Approximately one-third of patients may be asymptomatic. In children, the presentation is often less clearly respiratory in nature, with diarrhea, vomiting, failure to thrive and even cyanosis being more common . Symptoms may also be due to superadded opportunistic infections (see below). Signs include crackles on auscultation, clubbing or cyanosis.
Pathology
The understanding of PAP has evolved considerably over time.
PAP was originally defined by histopathologic findings of amorphous periodic acid-Schiff (PAS) stain (+) lipoproteinaceous material filling the alveoli, with relatively normal background lung architecture . Upon discovery that the PAS(+) material represents debris derived from the pulmonary surfactant, it became clear that PAP represents a disorder of surfactant turnover .
More recently, multiple causative factors for PAP have been elucidated. Alveolar macrophages, the key to surfactant homeostasis, are regulated by the granulocyte-macrophage-colony-stimulating factor (GM-CSF). Acquired autoimmunity to GM-CSF is now believed to be the most common cause of PAP in adults. Concurrent lung diseases which cause impaired alveolar macrophage function may also lead to a secondary form of PAP. Much more rarely, genetic mutations which phenotypically yield abnormal surfactant or GM-CSF cause a severe congenital form of PAP .
Thus, PAP may be divided into three broad categories:
- autoimmune: formerly referred to as idiopathic or primary, 90% of cases
- also termed adult or acquired
- IgG antibodies to GM-CSF
- secondary: 5-10% presents in individuals with other precipitating illness
- hematological malignancies
- inhalational lung disease
- silica (known as silicoproteinosis)
- titanium oxide
- immunodeficiency/immunosuppression (e.g. HIV/AIDS and after hematopoietic stem cell transplantation ) with coexistent infection, e.g. nocardiosis, aspergillosis, PCP
- congenital: 2%
- presents in the neonatal period in term babies
- poor prognosis if left untreated (lung transplantation)
- may be a distinct entity also known as chronic pneumonitis of infancy
- due to a mutation in genes encoding SP-B, SP-C, or GM-CSF receptor
Although imaging, bronchial lavage and sputum examination can strongly suggest the diagnosis, lung biopsy is sometimes required.
Markers
- elevated acute inflammatory markers, e.g. lactate dehydrogenase (LDH)
- (+) BAL or serum anti-GM-CSF antibodies
Radiographic features
As a general rule, radiographic features are often much more severe than the clinical presentation would suggest.
Plain radiograph
Chest radiograph findings are inconclusive . Findings can be variable, including :
- batwing pulmonary opacities:
- bilateral central symmetrical lung opacities with relative apical and costophrenic angle sparing
- reminiscent of pulmonary edema
- most common appearance in adults
- diffuse small pulmonary opacities
- reminiscent of a miliary pattern
- more common in children
- extensive diffuse consolidation
- reticulonodular opacities
Pleural effusions, cardiomegaly and lymphadenopathy are usually not features of uncomplicated PAP.
CT
Two main features characterize the appearance of pulmonary alveolar proteinosis on HRCT:
The combination of these two features is termed a crazy paving pattern, which although highly characteristic, is not pathognomonic. Additionally, pulmonary consolidation may be evident.
Lung changes are of either patchy or geographic distribution and may have a slightly lower lobe predilection .
Ground glass opacity typically resolves after therapeutic bronchoalveolar lavage, although septal thickening may persist.
Findings of pulmonary fibrosis are not typical of adult PAP , although are more common in neonatal disease .
Treatment and prognosis
Standard treatment includes therapeutic whole-lung bronchoalveolar lavage to remove alveolar material, although its role in children is less certain . GM-CSF supplementation has been used with varying effectiveness .
Prognosis is variable, ranging from improvement (with treatment) to a chronic and terminal course. A 30% 2-year mortality has been reported in adults prior to routine use of bronchoalveolar lavage . The 5-year mortality has now been reduced to approximately 5% . In children, that figure is much higher due to the reduced efficacy of bronchoalveolar lavage .
Double lung transplant has been used in the treatment of PAP, although the disease may recur .
Complications
- superimposed infection: especially with Nocardia asteroides sp. , although there are many others
- Aspergillus spp
- Candida spp
- Cryptococcus neoformans
- cytomegalovirus (CMV)
- Histoplasma capsulatum
- Mycobacterium (tuberculous and non-tuberculous)
- Pneumocystis spp
- Streptococcus pneumoniae
- pulmonary fibrosis (occurs in ≈30%)
Differential diagnosis
Imaging differential considerations for specific patterns include:
- crazy paving pattern
- ground glass opacities
- miliary opacities
- batwing pulmonary opacities
- pulmonary edema: pleural effusions and cardiomegaly usually absent in PAP
Siehe auch:
- Lungenödem
- Milchglasverschattungen
- miliare Lungenherde
- crazy paving-Muster
- diffuse Alveolarblutung
- Schmetterlingskonfiguration von Lungenverdichtungen
- Aussparung des subpleuralen Raums
und weiter:


 Assoziationen und Differentialdiagnosen zu Alveolarproteinose:
Assoziationen und Differentialdiagnosen zu Alveolarproteinose:




