Morbus Gaucher






Gaucher disease (GD) is the most common lysosomal storage disorder in humans. It is an autosomal recessive, multisystem disease arising from a deficiency of glucocerebrosidase or beta-glucosidase activity, resulting in the accumulation of a glycolipid (glucocerebroside) within the lysosomes of macrophages, particularity in the bone marrow, spleen and liver.
Epidemiology
Type 1 is the most common, affecting 1:500-1,000 Ashkenazi Jews and 1:50,000-100,000 of the general population . Types 2 and 3 are considered much rarer.
Clinical presentation
Age of presentation depends on the type of Gaucher disease:
- type 1 (most common form)
- age of presentation varies widely, with the mean age of diagnosis being 21 years of age
- some patients present in childhood while others remain asymptomatic throughout life
- clinical presentation tends to be with skeletal symptoms (bone pain, pathological fractures, osteonecrosis and bone crises ) , hepatomegaly, splenomegaly, and hematological disturbances
- type 2: evident by 6 months of age, with progressive neurological deterioration resulting in death by the age of 1 or 2
- type 3: presents with mild neurological complications by late adolescence or early childhood
Pathology
Genetic changes
The glucosylceramide beta (GBA) gene provides instructions for making ß-glucocerebrosidase. Mutations in the GBA gene reduce or eliminate the function of this lysosomal enzyme leading to a build-up of toxic glucocerebroside and related substances in various tissues and organs .
Classification
Three types of Gaucher disease are described, each with different manifestations :
- type 1 (non-neuropathic form or adult form): commoner type; progressive hepatomegaly, splenomegaly, anemia and thrombocytopenia, and marked skeletal involvement; lungs and kidneys may also be involved, but the CNS is spared
- type 2 (acute neuropathic form or infantile form): severe progressive neurological involvement with death by 1 to 2 years of age; hepatomegaly, splenomegaly, is also present (usually evident by 6 months of age)
- type 3: type 1 with neurological involvement
Radiographic features
Plain radiograph
Skeletal involvement is seen in 70-100% of patients and primarily involves long bones (tibia, humerus, femur) as well as vertebrae. Ribs, hands and wrists, ankles and feet, and mandible may also be involved . Features of skeletal involvement include:
- osteopenia
- osteonecrosis
- pathological/crush fractures
- endosteal scalloping
- Erlenmeyer flask deformities
- H-shaped vertebrae
- paranasal sinus obliteration due to medullary expansion
MRI
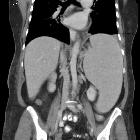
- spleen
- massive splenomegaly
- splenic nodules (30%)
- splenic infarcts (33%)
- liver
- hepatomegaly: less marked than the degree of splenomegaly
- T2: hyperintense stellate areas representing inflammation and fibrosis
- areas of hepatic ischemia
- skeletal system
- long bones are most severely affected
- reduced T1 and T2 signal from involved bone marrow (due to infiltration of Gaucher cells)
- bone marrow burden (BMB) score may be obtained from MRI images
- may give a "salt and pepper pattern" due to scattered involvement
- features of superimposed osteonecrosis
- metaphyseal notching of humeri
- pathological fractures
- Erlenmeyer flask deformity
Treatment and prognosis
Enzyme replacement with macrophage-targeted glucocerebrosidase has been shown to be highly effective in type 1 GD, halting the progression and even reversing both bone marrow and visceral infiltration . Radiographically, hepatomegaly and splenomegaly respond more rapidly than skeletal changes.
Glucosylceramide synthase inhibitors are available for patients with type 1 GD who cannot receive enzyme replacement therapy .
Complications
- osteonecrosis of the hip
- pathological fracture and pyogenic osteomyelitis
- lung involvement
- pulmonary infiltration by Gaucher cells (type 2)
- parenchymal infiltration with fibrosis (type 3)
- pulmonary hypertension
- increased frequency of multiple myeloma, Parkinson disease and Lewy body dementia
- Gaucheromas: rare pseudotumors comprising a mass of Gaucher cells
History and etymology
First described by French physician Philippe CE Gaucher (1854-1918) in 1882, while still a medical student .
Siehe auch:
- endosteal scalloping
- Aseptische Knochennekrose
- Splenomegalie
- Milzinfarkt
- Hämochromatose
- Auftreibung Metaphysen
- Hepatomegalie
- H-förmige Wirbel
- lysosomale Speicherkrankheit
und weiter:
- diffus hypointenses Knochenmarksignal in T1
- avascular necrosis causes (mnemonic)
- Platyspondylie
- Aseptische Wirbelkörpernekrose
- Hydrops fetalis
- Café-au-lait-Fleck
- Vertebra plana
- Bürstenschädel
- conditions involving skin and bone
- Knochen-in-Knochen-Aspekt
- Erlenmeyer flask deformity of the femur
- fetal splenomegaly
- Speicherkrankheit
- Sphingolipidose
- step off vertebrae
- Sichelzellenanämie Wirbelsäule


 Assoziationen und Differentialdiagnosen zu Morbus Gaucher:
Assoziationen und Differentialdiagnosen zu Morbus Gaucher: