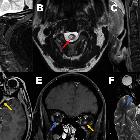
Opticusgliom









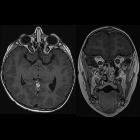



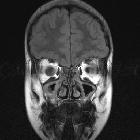




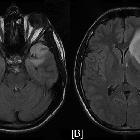
 nicht verwechseln mit: hypothalamisches Hamartom
nicht verwechseln mit: hypothalamisches HamartomOptic pathway gliomas are relatively uncommon tumors, with a variable clinical course and usually seen in the setting of neurofibromatosis type I (NF1). Histologically the majority are pilocytic astrocytomas.
They are characterized by imaging by an enlarged optic nerve seen either on CT or MRI. Usually showing low T1 and a high central T2 signal on MRI images, enhancement is variable.
Terminology
These tumors have sometimes been divided into optic pathway gliomas and hypothalamic gliomas (not to be confused with hypothalamic hamartomas). In cases where a tumor is confined to the optic nerves (Dodge stage 1 – see below), they can safely be referred to as optic nerve gliomas. Often, however, they are either centered on or extend to involve the chiasm and optic radiations. In such cases, they are difficult to distinguish from hypothalamic gliomas and such a distinction is in most instances artificial. In such more posterior cases, the term hypothalamic-optochiasmatic glioma is perhaps more accurate although it certainly does not roll off the tongue.
As such, generally, the term optic pathway glioma is favored, recognizing that there may be involvement of the hypothalamus.
Epidemiology
Optic pathway gliomas typically present in children, accounting for 10-15% of supratentorial tumors in this age group, and are often in the setting of neurofibromatosis type 1 (NF1) (10-63%) . In this setting, the tumors are often low-grade and indolent.
Males and females are approximately equally affected.
In adults, optic nerve gliomas do occur but are very rare and usually aggressive tumors . In such cases, no association with NF1 has been found .
Clinical presentation
Clinical presentation varies depending on the extent and location of the tumor.
Decreased vision (63%) is usually evident and can be documented with visual field examination if the child is old enough. In orbital optic nerve gliomas, eventually, mass effects will also occur with proptosis.
Involvement of the hypothalamus may result in polyuria/polydipsia , as well as obesity, sexual precocity and endocrine dysfunction (e.g. short stature). Diencephalic symptoms include a change in alertness and hyperactivity.
In large intracranial tumors, symptoms of raised intracranial pressure, focal neurological deficits and hydrocephalus from distortion of the midbrain may also be encountered.
Pathology
Most optic pathway gliomas are pilocytic astrocytomas (WHO grade I), although their imaging characteristics are not specific with regard to their histologic features. Varying degrees of cystic change and enhancement are demonstrated. The tumors may appear smooth, fusiform, eccentric, or lobulated.
Classification
The most widely accepted classification of optic pathway gliomas is the Dodge classification that was first proposed by Dodge et al. in 1958 . The Dodge classification divides these tumors into just three groups based on anatomical localization:
- stage 1: optic nerves only
- stage 2: chiasm involved (with or without optic nerve involvement)
- stage 3: hypothalamic involvement and/or other adjacent structures
A more recent modification of the Dodge classification has been proposed which further subdivides each stage .
Radiographic features
Plain radiograph
Radiographs no longer have any real role to play in the diagnosis of orbital masses, however, if performed enlargement of the optic canal may be demonstrated if a tumor is not confined to the orbit. Additional findings of neurofibromatosis type 1 may also be visible.
CT
CT is often the first investigation performed and although not as sensitive as MRI, the diagnosis can often be made, especially if thin slice imaging through the orbits is performed, or coronal and sagittal reconstructions obtained from volumetric data.
- enlargement of the optic nerve and the mass may either be fusiform or exophytic in appearance.
- enlargement of the optic canal
- the optic nerve may be elongated with kinking or buckling
MRI
MR imaging is optimal for showing the relationship of the mass to the hypothalamus, optic chiasm, and infundibulum as well as the intraorbital and intercanalicular components of the mass. Large tumors are typically heterogeneous with cystic and solid components.
- T1: enlargement, often iso to hypointense compared to the contralateral side
- T2
- hyperintense centrally
- thin low-signal at the periphery representing the dura
- T1 C+ (Gd): enhancement is variable
Treatment and prognosis
These tumors demonstrate variable clinical and radiological progression. In patients with NF1, it is not unusual for these tumors to be quiescent, with little progression demonstrated over some years. In others, the tumors are more aggressive with extension along the optic pathways .
Treatment options, therefore, depend on the clinical context, as well as the location of the tumor at presentation. If it is isolated to one optic nerve and does not extend to the chiasm, then resection is curative (albeit with the loss of vision in that eye). If a tumor extends to the chiasm or more posteriorly, then curative resection is not possible, with resection reserved for treatment of mass effects (proptosis, intracranial mass effect) .
Differential diagnosis
The main differential is that of optic nerve meningioma, however, the potential list is much longer including most causes of optic nerve enlargement.
The absence of calcification can be used to differentiate optic nerve glioma from optic nerve sheath meningioma .
Additionally, when the bulk of a tumor is located at the chiasm, the differential should include pituitary region masses.
Siehe auch:
- Pilozytisches Astrozytom
- Tumoren der Hypophysenregion
- intraorbitale Raumforderungen
- Neurofibromatose Typ 1
- Neuritis nervi optici
- hypothalamisches Hamartom
- Meningeom Nervus opticus
- hypothalamic lesions
- Gliom
- verdickter Nervus Opticus
- bilateral optic nerve glioma, Neurofibromatosis type 1
- Tumoren des Nervus opticus
- optic nerve glioma: neurofibromatosis type 1
und weiter:
- Makroadenom Hypophyse
- Neurosarkoidose
- Chiasma opticum
- verdickter Hypophysenstiel
- Pituitary MRI (an approach)
- Nervus opticus
- Metastasen in der Orbita
- oculomotor nerve palsy
- tram-track sign
- orbital pathology
- beschleunigte Skelettreifung
- Pituizytom
- intraokulare Raumforderungen
- Pilozytisches Astrozytom des Nervus opticus


 Assoziationen und Differentialdiagnosen zu Opticusgliom:
Assoziationen und Differentialdiagnosen zu Opticusgliom: