neurofibromatosis type 1






































Neurofibromatosis type 1 (NF1), also known as von Recklinghausen disease, is a multisystem neurocutaneous disorder, the most common phakomatosis, and a RASopathy. Additionally, it is also one of the most common inherited CNS disorders, autosomal dominant disorders, and inherited tumor syndromes.
Individual systemic manifestations are discussed individually:
- breast manifestations of NF1
- central nervous system manifestations of NF1
- cutaneous manifestations of NF1
- musculoskeletal manifestations of NF1
- pulmonary manifestations of NF1
- orbital manifestations of NF1
The remainder of this article pertains to a general discussion of neurofibromatosis type 1.
Epidemiology
Neurofibromatosis affects 1:2500-3000 individuals . In half of the cases, the disease is inherited as an autosomal dominant condition. In the other half, the disease is due to a de novo mutation . There is a variable expression but 100% penetrance by 5 years of age .
Clinical presentation
As is the case with many phakomatoses, NF1 results in a variety of abnormalities of variable severity. To make the clinical diagnosis two or more of the following are required :
- >6 cafe au lait spots evident during one year (prepubertal >0.5 cm, postpubertal >1.5 cm in size)
- two or more neurofibromas or one plexiform neurofibroma
- optic nerve glioma
- distinctive osseous lesion (such as sphenoid wing dysplasia or thinning of long bone cortex with or without pseudoarthrosis)
- two or more iris hamartomas (Lisch nodules)
- axillary or inguinal freckling
- a primary relative with NF1 with above criteria
A mnemonic to help remember these features is CAFE SPOT.
In addition, ~45% (range 30-60%) of patients have learning disabilities, and approximately 1% have hypertension due to renal artery stenosis.
It is important to consider that NF1 has a much earlier age of onset than schwannomatosis and NF2, with approximately 50% of patients meeting the diagnostic criteria for NF1 by the age of 1 year and approximately 97% meeting the criteria by the age of 8 years .
Neoplasms
It should come as no surprise that a disease due to inactivation of a tumor suppressor gene (see below) is also associated with an increased incidence of numerous tumors :
- pheochromocytoma
- malignant peripheral nerve sheath tumor (MPNST) ( ~10% of patients)
- Wilms tumor
- rhabdomyosarcoma
- renal angiomyolipoma
- glioma
- carcinoid tumor(s)
- leiomyoma(s)
- leiomyosarcoma
- ganglioglioma
- leukemia
Pathology
The NF1 gene locus is on chromosome 17q11.2 and the gene product is neurofibromin, which acts as a tumor suppressor of the Ras/MAPK pathway; inactivation of the gene thus predisposes to tumor development . For this reason, the disorder is classified as a RASopathy .
The disease primarily is a hamartomatous disorder that involves the ectoderm and mesoderm. Usually, three types of neurofibromas occur in this disorder and are distinguished on the basis of their gross and microscopic appearances.
- localized neurofibroma (cutaneous neurofibroma): the most common type, is a focal lesion that typically is located in the dermis and subcutis
- diffuse neurofibroma (subcutaneous neurofibroma): localized in the subcutis, usually in the head and neck region.
- plexiform neurofibroma: considered pathognomonic if present; they may be seen in virtually any location but usually occur in the neck, pelvis, and extremities
Radiographic features
Breast
Central nervous system
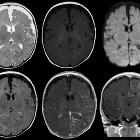
- FASI (focal areas of signal intensity): occur in deep white matter and basal ganglia or corpus callosum , areas of T2/FLAIR hyperintensity with no contrast enhancement
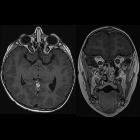
- optic nerve glioma or optic pathway glioma (may manifest as enlarged optic foramen)
- progressive sphenoid wing dysplasia
- lambdoid suture defects
- dural calcification at the vertex
- moya-moya phenomenon (rare)
- buphthalmos
Cutaneous
- cutaneous and subcutaneous neurofibromas: benign peripheral nerve sheath tumors
Skeletal
- kyphoscoliosis
- posterior vertebral scalloping
- hypoplastic posterior elements
- enlarged neural foramina
- ribbon rib deformity, rib notching, and dysplasia
- dural ectasia
- tibial pseudoarthrosis or, less commonly, ulnar pseudoarthrosis
- bony dysplasias: especially affecting tibia
- severe bowing, gracile bones
- multiple non-ossifying fibromas
- limb hemihypertrophy
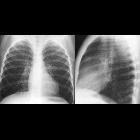
Thoracic
- mediastinal masses
- neurofibroma
- lateral thoracic meningocele: typically on the convex side of scoliosis (through widened neural foramina)
- extra-adrenal pheochromocytoma
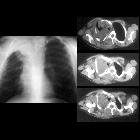
- lung parenchymal disease: ~20%
- diffuse interstitial fibrosis: lower zone
- bullae formation: upper zone
- secondary pulmonary arterial hypertension and cor pulmonale
Vascular
Treatment and prognosis
No single treatment exists, and a combination of supportive and surgical therapies are employed depending on the specific tumors and anomalies present.
Although prognosis is very variable, overall life expectancy is approximately half that of non-affected individuals. Tumors or cardiovascular complications are the most common causes of mortality .
History and etymology
The first name of this condition was Von Recklinghausen disease because, in 1882, von Recklinghausen described cases of neurofibromatosis and recognized it as a nosological entity .
See also
Siehe auch:
- Rippenusuren
- Pilozytisches Astrozytom
- Angiomyolipom der Niere
- Arteriovenöse Malformation
- Aortenisthmusstenose
- Aneurysma
- Gangliogliom
- Karzinoid
- Rhabdomyosarkom
- Nierenarterienstenose
- diffuses intrinsisches Ponsgliom
- maligner peripherer Nervenscheidentumor (MPNST)
- pulmonale Hypertonie
- Phäochromozytom
- Leiomyosarkom
- Opticusgliom
- scalloping Wirbelkörper
- Neurofibrom
- Duraektasie
- Nephroblastom
- Phakomatosen
- cafe-au-lait spots
- spinales Pilozytisches Astrozytom
- spinales Astrozytom
- Moyamoya-Muster
- pulmonale Bullae
- plexiformes Neurofibrom
- Orbitadysplasie
- neurofibromatosis of the breast
- different types of neurofibroma
- localized neurofibroma
und weiter:
- Fibröse Dysplasie
- Skoliose
- basal ganglia T1 hyperintensity
- Café-au-lait-Fleck
- multiple zystische Lungenherde
- Staphylom
- BIRADS II
- Moyamoya-Erkrankung
- Makrozephalie
- macrodystrophia lipomatosa
- Neurofibromatose Typ 2
- sphenoid wing dysplasia
- Tibiaverbiegung bei Kindern
- Medulläres Schilddrüsenkarzinom
- Angiosarkom Leber
- dural ectasia of lumbo-sacral spine
- stroke in children and young adults
- Neurofibromatose
- Verbiegung der langen Röhrenknochen
- intradural spinal mass lesions - an approach
- vergrößerter Bulbus oculi
- renovaskuläre arterielle Hypertonie
- Hemihyperplasie
- congenital pseudoarthrosis
- multiple benign lucent bone lesions (mnemonic)
- bilateral megalencephaly
- Erweiterung des interpedunculären Abstands
- Makrodaktylie
- radial pseudoarthrosis (congenital)
- Grisel-Syndrom
- Neurocristopathien
- Typen von Neurofibromen
- fetal arachnoid cyst
- Kongenitale Pseudarthrose der Tibia
- diffuse neurofibroma
- kongenitale Pseudarthrose der Fibula
- localised neurofibroma
- Triton-Tumor
- kongenitale Ulnapseudarthrose
- bilateral optic nerve glioma, Neurofibromatosis type 1
- Neurofibromin
- Neurofibromatose Typ 1 und Neurofibrosarkom
- Nervenscheidentumor
- Megaloenzephalie
- optic nerve glioma: neurofibromatosis type 1
- neurofibromatosis type 1 associated with a neurofibrosarcoma of the urinary bladder
- neurofibromatosis type 1: orbital manifestations
- lokaler Gigantismus
- intracranial findings in neurofibromatosis type 1
- diffusion MRI of myelin vacuolization in NF1
- mesenteric plexiform neurofibroma
- Neurofibromatose Typ 1 und Pseudarthrose der Ulna
- neurofibromatosis I affecting long bones
- neurofibroma of the ankle
- bare orbit sign


 Assoziationen und Differentialdiagnosen zu Neurofibromatose Typ 1:
Assoziationen und Differentialdiagnosen zu Neurofibromatose Typ 1: