Pleomorphes Xanthoastrozytom








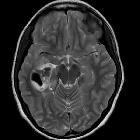








Pleomorphic xanthoastrocytomas (PXA) are a type of rare, low-grade astrocytoma (WHO Grade II) found in young patients who typically present with temporal lobe epilepsy.
They usually present as cortical tumors with a cystic component and vivid contrast enhancement. Features of slow growth may be present, such as no surrounding edema and scalloping of the overlying bone. A reactive dural involvement expressed by a dural tail sign can be found. Calcifications are rare.
Epidemiology
They are rare tumors accounting for only 1% of primary brain tumors . Typically these tumors are found in young patients (children or young adults), with a peak incidence in the second and third decade of life (10-30 years) .
Clinical presentation
As these tumors have a predilection for the temporal lobe, they most frequently present with seizures (~75% of cases) . Other findings include dizziness, and headache or rarely patients are asymptomatic .
Pathology
Grading
Pleomorphic xanthoastrocytomas are considered WHO grade II tumors. If mitoses are more frequent (>5 mitoses per 10 high-power fields) then a diagnosis of anaplastic pleomorphic xanthoastrocytoma (WHO grade III) should be made .
Location
Pleomorphic xanthoastrocytomas are almost invariably (98%) located supratentorially, typically located superficially (peripherally) abutting the leptomeninges, involving the cortex and overlying leptomeninges but actual dural involvement is rare. Approximately half are located in the temporal lobe, with the remainder of the lesions more common in the frontal than the parietal lobes .
Macroscopic appearance
Macroscopically these tumors appear well-circumscribed, often with a cystic component and involvement of the overlying leptomeninges .
Microscopic appearance
Microscopically the margins are not as well defined. The histological features are variable (thus the term 'pleomorphic') with spindle cells, polygonal cells, multinucleated cells and lipid-laden xanthomatous astrocytes are all identified . Even more pleomorphic is the appearance of the nuclei, with common nuclear inclusions, and highly variable nuclear size .
Immunophenotype
Immunohistochemistry demonstrates expected glial marker reactivity. Less obviously, there is also variable reactivity for neuronal markers :
- GFAP: positive, although often only weakly
- S100: positive
- neuronal markers including synaptophysin, MAP2 and neurofilament: variable
Ki-67 proliferation index: <1%
Genetics
Pleomorphic xanthoastrocytomas, as well as pilocytic astrocytomas (and many non-CNS tumors), exhibit BRAF mutations . The only reported association is with neurofibromatosis type 1, although this is not a strong association .
Radiographic features
Pleomorphic xanthoastrocytomas appear as a solid enhancing nodule, frequently with a peripheral eccentric cystic component (50-60%). Due to their peripheral location and leptomeningeal involvement, they are one of the tumors that may exhibit a dural tail. This is reactive rather than due to true direct dural invasion, which is rare . As these lesions are very slow growing, superficial remodeling of the adjacent skull is characteristic and vasogenic edema is variable .
CT
Pleomorphic xanthoastrocytomas are typically hypodense or isodense and may be well or poorly demarcated, usually with little or no surrounding edema. Calcification is rare. Due to its superficial location it may cause scalloping of the overlying bone .
MRI
- T1
- solid component iso to hypointense cf. grey matter
- cystic component low signal
- leptomeningeal involvement is seen in over 70% of cases
- T1 C+ (Gd)
- solid component usually enhances vividly
- T2
- solid component iso to hyperintense cf. grey matter
- cystic component high signal
- on T2 FLAIR sequence, cystic areas show hyperintensity relative to CSF due to higher protein contents
- little surrounding vasogenic edema
DSA - angiography
Despite vivid enhancement, pleomorphic xanthoastrocytomas are usually avascular on angiography
Treatment and prognosis
Although prognosis is good following surgical excision, with a 5-year survival of 90% and 5-year-disease-free-survival of 70% , local recurrence and malignant transformation (to WHO grade III lesion or GBM) are common, encountered in up to 20% of cases .
Neither radiotherapy nor chemotherapy has a significant effect on these tumors , although radiotherapy may have a role to play in patients with incomplete resection or those with recurrent disease .
Differential diagnosis
Main differential diagnosis is that of other cortical tumors, with helpful distinguishing features including :
- ganglioglioma
- can look very similar
- contrast enhancement often less prominent
- calcification in ~50% of cases
- no dural tail sign
- dysembryoplastic neuroepithelial tumors (DNET)
- contrast enhancement uncommon
- 'bubbly appearance' common
- oligodendroglioma
- calcifications common
- desmoplastic infantile ganglioglioma
- young children
- dural involvement prominent
- large often multiple lesions
- cystic meningioma
Siehe auch:
- Pilozytisches Astrozytom
- Oligodendrogliom
- Gangliogliom
- Astrozytom
- Dysembryoplastischer neuroepithelialer Tumor
- Dural-Tail-Zeichen
- Hirntumoren
- WHO Grade II
- temporal lobe masses
- temporal lobe epilepsy.
und weiter:


 Assoziationen und Differentialdiagnosen zu Pleomorphes Xanthoastrozytom:
Assoziationen und Differentialdiagnosen zu Pleomorphes Xanthoastrozytom:





