Alpha-1-Antitrypsin-Mangel
















Alpha-1-antitrypsin (A1AT) deficiency is a hereditary metabolic disorder and is the most common genetic cause of emphysema and metabolic liver disease in children. It results in the unopposed action of neutrophil elastase and subsequent severe basal panlobular emphysema and respiratory symptoms. Accumulation of altered alpha-1-antitrypsin in hepatocytes incites an inflammatory response and chronic liver disease.
Epidemiology
Estimated prevalence is thought to be around 1 in 1,500 to 3,500 individuals with European ancestry. It reported to be uncommon in people of Asian descent.
Clinical presentation
The classic presentation of the disease is with dyspnea in the 4 to 5 decades of life. Two-thirds of individuals show clinical features but most carriers are asymptomatic.
Pathology
Alpha-1-antitrypsin (A1AT) is a protein that prevents enzymes such as elastase from degrading normal host tissue. Over 90% of the alpha-1-antitrypsin protein is produced in hepatocytes by codominant gene expression on chromosome 14. The alpha-1-antitrypsin protein inhibits neutrophil elastase. In patients with severe deficiency, the neutrophil elastase acts unopposed resulting in damage to the lower respiratory tract. This damage is predominantly basal because of the gravitational distribution of pulmonary blood flow.
Associations
- asthma
- pancreatitis
- aneurysms, including intracranial aneurysms
Radiographic features
Thoracic manifestations
Plain radiograph and CT
-
- the emphysema pattern was traditionally thought to be panlobular, although more recent studies have also suggested a variable pattern to the emphysema
- basal predominance, in contradistinction to centrilobular emphysema, which shows apical predominance; the two conditions can coexist in smokers
- emphysema may develop in 75-85% of cases
- bronchiectasis: up to 40%
- frank bullae formation: non-specific feature
- bronchial wall thickening: non-specific feature
- hepatopulmonary syndrome
Nuclear medicine
Reduced perfusion and ventilation in the lower zones on a ventilation/perfusion (V/Q) study.
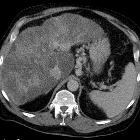
Abdominal manifestations
Hepatic manifestations of this disease are those of cirrhosis.
Treatment and prognosis
Emphysema and cirrhosis are usually considered the most common causes of death .
Survival is substantially worse in smokers, who have a 20-year decrease in longevity relative to non-smokers. According to one study, the overall median survival time was ~55 years .
Alpha-1-antitrypsin replacement therapy, most often by weekly intravenous infusions of alpha-1-antitrypsin purified from human plasma, has been used in some situations to partially correct the biochemical defect .
While randomized, placebo-controlled clinical trials have confirmed a reduction in the decline in lung density in patients receiving augmentation therapy , its efficacy in reducing mortality is uncertain . Other management strategies include avoidance of smoking and of other risk factors for cirrhosis.
Lung and liver transplant is generally reserved for those with end stage disease .
Complications
- cirrhosis with increased risk of hepatocellular carcinoma
- A1AT deficiency carriers are at 70-100% increased risk of lung cancer
Differential diagnosis
The differential will depend on the organ involved:
- for thoracic manifestations: see differential for panlobular emphysema
- near identical imaging appearances with ritalin lung
- for hepatic manifestations: see differential for cirrhosis
Siehe auch:
- Bronchiektasen
- Leberzirrhose
- panlobuläres Lungenemphysem
- hepatozelluläres Karzinom
- Hepatopulmonales Syndrom
- pulmonale Bullae
und weiter:


 Assoziationen und Differentialdiagnosen zu Alpha-1-Antitrypsin-Mangel:
Assoziationen und Differentialdiagnosen zu Alpha-1-Antitrypsin-Mangel: