Huntington's disease












Huntington disease (HD), also known as Huntington chorea, is an autosomal dominant trinucleotide repeat neurodegenerative disease characterized by a loss of GABAergic neurons of the basal ganglia, especially atrophy of the caudate nucleus and putamen (dorsal striatum). Huntington disease is clinically characterized by progressive unintentional choreoathetoid movements, subcortical type dementia, behavioral changes, and psychosis which starts in midlife.
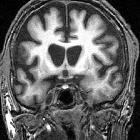
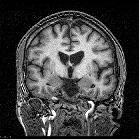
On imaging, it is classically characterized by atrophy of the caudate nucleus with concomitant enlargement of the frontal horns of the lateral ventricles.
Epidemiology
Huntington disease has a prevalence of 5-10 per 100,000 and is typically diagnosed between 30 and 50 years of age . Incidence is equal in both genders, although there appears to be an effect depending on the gender of the parent from whom the defect was inherited: if inherited from the father, presentation is earlier. The cause for this effect is as yet uncertain .
In approximately 1-6% of patients, symptoms occur before the age of 20 years, so-called 'juvenile' form, which appears to be a variant of the usual adult form, with a different pattern of symptoms. In juvenile cases having inherited the disease from the father is far more common .
Clinical presentation
Presentation is typically with progressive rigidity, choreoathetosis, dementia, psychosis, and emotional lability .
The juvenile form has a different presentation, with cerebellar symptoms, rigidity, and hypokinesia being prominent.
Pathology
Genetics
The inheritance pattern of Huntington disease is autosomal dominant with complete penetrance and genetic anticipation (i.e. next generation will have more repeats of CAG and a more severe course of the disease or show symptoms earlier) particularly if the inherited mutated allele is paternal. The mutation responsible is on chromosome 4p16:3 and consists of a CAG trinucleotide repeat. The usual 10-30 copies are amplified to >36, and the greater the number of repeats the earlier the age of onset .
Microscopically, there are Huntington nuclear inclusion bodies . Both deep grey matter and, to a lesser degree, white matter are involved in HD.
Radiographic features
Although all modalities capable of structural brain imaging will demonstrate morphological changes of Huntington disease, MRI has the greatest spatial and contrast resolution and is thus preferred.
MRI
The most striking and best-known feature is that of caudate head atrophy. There is also, however, prominent putaminal volume loss which is usually not as easily recognized on visual inspection but seen well on morphometry . This is particularly the case in younger patients . The combination results in enlargement of the frontal horns, often giving them a "box" like configuration .
This can be quantified by a number of measurements:
- frontal horn width to intercaudate distance ratio (FH/CC)
- intercaudate distance to inner table width ratio (CC/IT)
In some cases, the basal ganglia may show decreased T2 signal and blooming on SWI in keeping with iron deposition . Generalized age-inappropriate cortical volume loss is also recognized .
MR spectroscopy may demonstrate elevation of lactate in the occipital cortex and basal ganglia which correlates with duration of symptoms. There is also a decrease in NAA/creatine ratio in keeping with neuronal loss in the basal ganglia.
PET/CT
PET scan demonstrates hypometabolism by decreased FDG uptake in basal ganglia and frontal cortex even before noticeable caudate nucleus volume loss .
Treatment and prognosis
No treatment is currently generally available .
The adult-onset form is slower in its course and inevitably leads to death in 14-15 years, whereas the juvenile form has a more rapidly progressive course, with death occurring in 7-8 years .
History and etymology
The condition is named after George Huntington (1850-1916), an American physician .
Differential diagnosis
- Wilson disease: more commonly involves white matter, thalamus, cerebellum and brainstem
- Leigh disease: more commonly involves white matter, thalamus, cerebellum and brainstem
- Hallervorden-Spatz syndrome
- acute hypoxic encephalopathy
- carbon monoxide poisoning
- hypoglycemic encephalopathy
Siehe auch:
- Putamen
- Morbus Wilson
- Leigh-Syndrom
- Basalganglien
- Kohlenmonoxidintoxikation
- Neurodegeneration mit Eisenablagerung im Gehirn
- Nucleus caudatus
- Atrophie caput nuclei caudati
und weiter:


 Assoziationen und Differentialdiagnosen zu Chorea Huntington:
Assoziationen und Differentialdiagnosen zu Chorea Huntington:




