Morbus Alexander



Alexander disease, also known as fibrinoid leukodystrophy, is a rare fatal leukodystrophy, which usually becomes clinically evident in the infantile period, although neonatal, juvenile and even adult variants are recognized. As with many other diseases with variable age of presentation, the earlier it manifests the more fulminant the clinical course.
There are three clinical forms:
Childhood-onset Alexander disease
Childhood-onset Alexander disease is sporadic and typically presents with macrocephaly, rapid neurological deterioration, seizures and spasticity, and retarded psychomotor development.
In some cases, the gene for glial fibrillary acidic protein (GFAP) has been implicated. Histologically the disease is characterized by the accumulation of large numbers of Rosenthal fibers and eosinophilic granular bodies.
Clinical presentation
It generally presents in infants and adolescents. Macrocephaly is typically present, and other clinical features include progressive quadriparesis and intellectual failure.
Pathology
Most of the cases are sporadic. However, familial disease has also been reported. A heterozygous mutation in the coding region, mapped to chromosome 17q21, of GFAP an astrocyte-specific intermediate filament protein, is associated with most cases of infantile sporadic onset.
Histologically the disease is characterized by the accumulation of large numbers of Rosenthal fibers and eosinophilic granular bodies (large accumulations agglomerations of astrocytic processes) in the degenerated (demyelinated) white matter which is a product of GFAP. This is on its own a non-specific finding, as they are also seen in slow-growing or benign astrocytomas (e.g. pilocytic astrocytomas). Intracellular deposition of Rosenthal fibers may cause abnormal functioning of the oligodendrocytes.
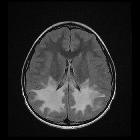
Radiographic features
The disease begins in the frontal region and extends posteriorly. Subcortical U-fibers are somewhat initially spared but affected relatively early in the course of the disease. End-stage disease is characterized by contrast-enhancing cystic leukomalacia.
MRI
- T2: increased signal in
- bifrontal white matter which tends to be symmetrical
- caudate head > globus pallidus > thalamus > brain stem
- periventricular rim
- T1 C+ (Gd): enhancement may be seen in the same areas
Obstructive hydrocephalus secondary to periaqueductal involvement and swelling of basal ganglia may be seen.
Juvenile/adult-onset Alexander disease
Adult-onset Alexander disease (AOAD), which was only recognized with any frequency after GFAP was recognized as the mutation, has markedly different imaging findings and presentation. It has been described as autosomally inherited in some pedigrees, presenting with prominent bulbar symptoms (dysphagia, dysphonia or dysarthria), sleep disturbance and impairment of autonomic function.
Radiographic features
MRI
- T2: increased T2 signal (demyelination) and atrophy of the
- medulla and upper cervical cord
- occasional involvement of inferior cerebellar peduncles
- pons, middle and superior cerebellar peduncles (rare)
- supratentorial (typically periventricular) may be seen in patients <40 years of age
- T1 C+ (Gd): patchy enhancement uncommonly seen in adult patients
Siehe auch:
- Pilozytisches Astrozytom
- Verschlusshydrocephalus
- Makrozephalie
- Leukodystrophie
- rosenthal fibres
- juvenile Form des Morbus Alexander
- Morbus Alexander Kriterien nach van der Knaap
und weiter:


 Assoziationen und Differentialdiagnosen zu Morbus Alexander:
Assoziationen und Differentialdiagnosen zu Morbus Alexander: