Sturge-Weber syndrome






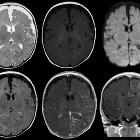
























Sturge-Weber syndrome, or encephalotrigeminal angiomatosis, is a phakomatosis characterized by facial port wine stains and pial angiomas.
It is part of a wide spectrum of possible phenotypes included in the craniofacial arteriovenous metameric syndrome (CAMS).
Epidemiology
Sturge-Weber syndrome is a rare syndrome, with an incidence estimated at 1 case in 20,000-50,000 persons .
Clinical presentation
The diagnosis is usually obvious on account of a congenital facial cutaneous hemangioma (also known as port wine stain or facial nevus flammeus). This feature is almost always present and usually involves the ophthalmic division (V1) of the trigeminal nerve; if this territory is not involved, Sturge-Weber syndrome is unlikely. In ~5%, it has intracranial involvement present without associated cutaneous nevus. In the majority of cases (~70%) the nevus is unilateral and ipsilateral to the intracranial abnormality.
The most common clinical manifestation is with childhood seizures, present in 71-89% of cases , which is often refractory to medical therapy. These usually begin in the first few years of life and are often associated with developmental delay and hemispheric symptoms including hemiplegia/hemiparesis and hemianopsia.
Approximately a third of patients have choroidal or scleral angiomatous involvement, which may be complicated with retinal detachment, buphthalmos or glaucoma .
Pathology
Unlike most phakomatoses, Sturge-Weber syndrome is sporadic with no definite identifiable hereditary component . An associated gene mutation has been identified with a nucleotide transition in GNAQ on chromosome 9q21 .
The leptomeningeal hemangioma results in a vascular steal affecting the subjacent cortex and white matter producing localized ischemia. In about 80% of the cases, there is unihemispheric involvement.
Associations
Classification
According to Roach et al . Sturge-Weber syndrome can be classified according to the presence/absence of facial and leptomeningeal angiomas:
- Type I: represents classic syndrome, with both facial and leptomeningeal angiomas; may have glaucoma.
- Type II: facial angioma without evidence of intracranial disease; may have glaucoma.
- Type III: isolated leptomeningeal angioma; usually no glaucoma.
Radiographic features
Plain radiograph
Skull x-rays were historically useful and capable of identifying the gyriform calcification of the subcortical white matter although they no longer play a significant role in the diagnosis or management of this condition. The finding usually becomes evident between 2 and 7 years of age .
CT
- detects subcortical calcification at an earlier age than plain film and can also demonstrate associated parenchymal volume loss
- tram-track sign of cortical and subcortical calcification
- calvarial and regional sinus enlargement may be evident
- ipsilateral choroid plexus may be enlarged
- in severe cases, a Dyke-Davidoff-Masson appearance may be seen
- orbital choroidal hemangiomas may be present
- asymmetric cavernous sinus enlargement
MRI
- T1: signal of affected region largely normal, with anatomic volume loss evident at older age
- T1 C+ (Gd)
- prominent leptomeningeal enhancement in affected area (due to congested internal cerebral veins, a manifestation of the so-called 'pial angiomatosis', resulting in venous congestive ischemia with infarction and obliteration of cerebral parenchyma)
- much later in life, the angioma may 'burn out' losing enhancement
- enlarged ipsilateral choroid plexus
- dilatation of transparenchymal veins that communicates between the superficial and deep venous systems
- T2
- low signal in white matter subjacent to angioma representing
- postulated accelerated myelination in neonate
- calcification later in life
- abnormal deep venous drainage seen as flow voids
- low signal in white matter subjacent to angioma representing
- GE/SWI/EPI: sensitive to calcification, seen as regions of signal drop out
- MR spectroscopy: decreased NAA
Angiography (DSA)
In most cases (82%), angiography is abnormal and demonstrates absent superficial cortical veins with abnormal and enlarged deep venous drainage .
Treatment and prognosis
Treatment revolves primarily around seizure control, with surgical resection only indicated rarely in refractory cases. Ophthalmological examination is also essential to identify and treat ocular involvement .
History and etymology
Sturge-Weber syndrome was first described by Sturge in 1879 who argued that there was a direct link between the intracranial hemangioma and the clinical presentation, although this was not accepted by his medical peers. It took until 1901 for Kalischer to provide the pathological confirmation that the pial angioma caused the neurological sequelae .
In 1912 Weber and Volland described the intracranial calcification. Radiographic identification of cerebral calcification was first described by Dimitri in 1922 . In fact, Schirmer described a male patient with a facial nevus and buphthalmos in 1860, however, he did not recognize that it was a neurological condition .
- William Allen Sturge: English physician (1850-1919)
- Frederick Parkes Weber: English dermatologist (1863-1962)
- five different diseases are named after Dr Weber!
- Vincente Dimitri: Austrian dermatologist (1885-1955)
- Rudolf Schirmer (1831–96): German ophthalmologist
- Siegfried Kalischer (1862-1954) German neuropathologist
Differential diagnosis
The differential is a combination of that for multiple intracranial calcifications, cerebral hemiatrophy and leptomeningeal enhancement, and therefore includes:
- cerebral arteriovenous malformation (AVM)
- infection
- PHACE syndrome
- healed cortical infarct
- radiotherapy
- Gobbi syndrome
Siehe auch:
- zerebrale Verkalkungen
- intrakranielle Verkalkungen
- Neurozystizerkose
- zerebrale arteriovenöse Malformation
- leptomeningeale Kontrastmittelanreicherungen
- einseitige Hirnatrophie
- TORCH
- F.P. Weber-Syndrom
und weiter:
- Developmental Venous Anomaly
- fetal intracranial calcification
- Brain stone
- neuroradiologisches Curriculum
- Dyke-Davidoff-Masson syndrome
- kongenitale vaskuläre Malformationen
- Phakomatosen
- angiomatosis
- Hemimegalencephalie
- Rasmussen-Enzephalitis
- Neurocristopathien
- syndromes with a vascular component
- Teleangiektasie
- leptomeningeale Verkalkungen
- cortical calcification
- Arteriovenöse Malformation des Gesichts
- vaskuläre Syndrome
- hemispherectomy
- Sturge Weber syndrome associated with pleomorphic xanthoastrocytoma
- tram-track calcifications


 Assoziationen und Differentialdiagnosen zu Sturge-Weber-Krabbe-Syndrom:
Assoziationen und Differentialdiagnosen zu Sturge-Weber-Krabbe-Syndrom:






