adrenocortical carcinoma
















Primary adrenal cortical carcinoma (also known as adrenocortical carcinoma) is a highly malignant but rare neoplasm. It may present as a hormonally active or an inactive tumor.
Epidemiology
Although men and women are affected equally, functioning tumors are more common in females, who are also more likely to have an associated endocrine syndrome . The median age of presentation is around 50 years, but tumors have been found at all ages .
An additional, but smaller, incidence peak occurs in early childhood. In these cases, tumors are more likely to be functioning .
Incidence is low, ranging from 0.6 to 1.67 per million, per year .
Clinical presentation
Hormonally inactive tumors present as palpable masses, abdominal pain or with evidence of metastasis. Hormonally active tumors, representing 30-40% of all adult tumors, present with characteristic clinical manifestations :
- Cushing syndrome secondary to elevated cortisol (most common)
- virilisation or feminization secondary to elevated androgens
- Conn syndrome secondary to hyperaldosteronism (rare)
Associations
- Beckwith-Wiedemann syndrome
- Li-Fraumeni syndrome
- Carney complex
- multiple endocrine neoplasia type 1
- familial adenomatous polyposis
Radiographic features
Ultrasound
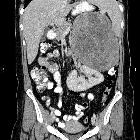
Often a suprarenal well-defined mass is seen, the appearance of which varies based on the size of the lesion. Smaller lesions are homogenous, whereas larger lesions are heterogeneous secondary to necrosis/hemorrhage.
CT
CT is usually the first imaging modality used.
- tend to be large (>6 cm)
- irregularly-shaped
- central areas of necrosis and hemorrhage, resulting in variable enhancement
- relative contrast retention (washout <40%) on delayed contrast-enhanced CT
- calcification is seen in up to 30% of cases
If the mass is found early, when still small, then it is difficult to distinguish from an adenoma, as aggressive features are often absent .
Focal extension into renal vein, IVC and liver are relatively common, with some series finding renal vein involvement in up to 40% of patients . Metastasis to regional lymph nodes, lung, bones, and liver can occur. Liver metastases tend to be hypervascular.
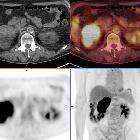
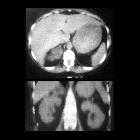
MRI
MRI can be useful to determine hepatic invasion if CT is inconclusive.
Heterogeneous mass is seen that is of high signal on T2 sequences. Areas of hemorrhage may result in variable signal intensity dependent on the age of the hemorrhage. Heterogeneous enhancement is seen with administration of gadolinium .
Angiography
Arteriography may be performed in patients who present with a large mass for which the organ of origin cannot be determined . Selective catheterization can identify the primary vascular supply, and thus help distinguish adrenal from renal tumors.
Treatment and prognosis
Ideally, treatment is with surgical excision, however, in many instances, the disease is advanced at the time of diagnosis, in which case chemotherapy (Mitotane) and radiation may be given for palliation.
In general, males have a poorer prognosis, largely because they are less likely to have functioning tumors, and thus present later, with more advanced disease .
Differential diagnosis
For an adrenal mass consider other adrenal lesions :
- adrenal metastases
- known primary neoplasm
- adrenal adenoma
- adrenal hemorrhage
- pheochromocytoma
- may look identical on imaging
- differentiation is on histology, biochemistry and functional status
Practical points
- percutaneous fine needle aspiration (FNA) of adrenal cortical carcinoma is unreliable for establishing the diagnosis
- the most specific indications of adrenal cortical carcinoma are metastases and local invasion
Siehe auch:
und weiter:
- Nebennierenraumforderungen
- Myelolipom Nebenniere
- genitourinary curriculum
- Nierenvenenthrombose
- Verkalkungen der Nebennnieren
- Gynäkomastie
- Beckwith-Wiedemann-Syndrom
- lipomatöse Impression der Vena cava inferior
- Myelolipom
- multiple endokrine Neoplasie Typ 1
- Budd-Chiari-Syndrom
- Tumorthrombus
- Hämangiom der Nebenniere
- Nebennierenrinden-Tumor
- Sarkome der Nebenniere
- myxoides Karzinom der Nebennieren
- myxoid adrenocortical carcinoma
- Nebennierenrindenkarzinom bei Kindern


 Assoziationen und Differentialdiagnosen zu Nebennierenrindenkarzinom:
Assoziationen und Differentialdiagnosen zu Nebennierenrindenkarzinom:

