glutaric aciduria type 1







Glutaric aciduria type 1 is a leukodystrophy that can be subclassified as an organic acidopathy. It has a highly variable clinical presentation, and laboratory investigations are not always diagnostic. Imaging, therefore, has an important role to play as the MRI features can be characteristic.
Epidemiology
Glutaric aciduria type 1 is a rare organic aciduria, with an estimated prevalence of 1 in 100,000 newborns . It is inherited in an autosomal recessive manner, and hence consanguineous marriages are a risk factor.
Clinical presentation
Glutaric aciduria type 1 has a variable presentation, but typically, affected neonates are asymptomatic in the first few months of life , other than developing macrocephaly. They tend to present with an acute encephalopathy following concurrent infection or acute catabolic state e.g. gastrointestinal disturbance. Hence, the initial presentation may resemble viral encephalitis or ADEM. Following the acute presentation, extrapyramidal symptoms develop which correlate, on imaging, with striatal involvement and subsequent necrosis .
A subset of patients present with an insidious onset without episodes of acute deterioration , and others still present as adults with progressive encephalopathy, or are asymptomatic . A wide range of presentation may thus be encountered.
Pathology
Glutaric aciduria type I is caused by an inherited deficiency of the enzyme glutaryl-CoA dehydrogenase. This leads to accumulation of glutaric acid and 3-hydroxyglutaric acid in the brain and body fluids, including urine (hence the name glutaric aciduria). Type II glutaric aciduria is a different disease caused by unrelated enzyme deficiencies .
Markers
Routine blood, urine and CSF analysis for the metabolites mentioned above may be misleading, as excretion is only intermittent even during episodes of acute decompensation . Imaging, therefore, has an important role to play in these situations.
Genetics
Definitive diagnosis of glutaric aciduria type 1 can be established by DNA-based analysis, looking for mutations in the GCDH gene on chromosome 19 .
Radiographic features
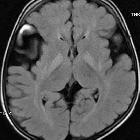
MRI is the modality of choice in the assessment of glutaric aciduria type 1. In clinically severely affected children, bilateral basal ganglia abnormalities are seen, with initial swelling that subsequently progresses to atrophy and necrosis. Other grey matter structures can be affected, e.g. the substantia nigra and the dentate nuclei. Hyperintensity of the tegmental tracts along the fourth ventricle floor has also been described. Delay in myelination is a further finding in severely affected infants.
The above changes are not necessarily seen in less severely affected children. Common features in both groups of patients are macrocephaly, expansion of subarachnoid convexity spaces, and wide CSF spaces anterior to the temporal poles and in the Sylvian fissures. Whether these wide CSF spaces represent arachnoid cysts rather than atrophy and under-opercularisation of the Sylvian fissures, remains unresolved.
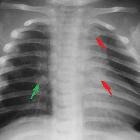
With the expansion of convexity subarachnoid spaces, the coursing bridging veins are susceptible to rupture with only minor trauma, and these patients may present with subdural hemorrhages. In this context, the radiologist needs to be familiar with the imaging findings of glutaric aciduria type I so that an erroneous diagnosis of non-accidental injury is not made. The findings described above are not by themselves specific, but the combination of these findings in a macrocephalic child with extrapyramidal symptoms are at least highly suggestive, if not pathognomonic .
Signal characteristics:
- T1: low signal
- T2/FLAIR: high signal
- GE/SWI: no susceptibility effect
- DWI: restricted diffusion acutely
- T1 C+ (Gd): no enhancement
- MR spectroscopy: lactate peak within basal ganglia acutely
Treatment and prognosis
Glutaric aciduria type 1 is a slowly progressive disease, with episodes of acute deterioration, often following infection. The progressive extrapyramidal symptoms are disabling, but mental capabilities may remain preserved . If untreated, death is usually in the first decade, in the setting of an acute exacerbation.
Early post-natal diagnosis should be sought as an early treatment prior to metabolic decompensation has the best chance of preventing neurological deterioration (once a metabolic crisis occurs, basal ganglia involvement is inevitable). For this reason, all siblings of an affected child and all future pregnancies should be screened for the disease. Nevertheless, some cases are progressive despite all appropriate treatment .
Treatment in the acute stage takes the form of prevention and correction of the catabolic state . In the chronic stage, a low protein diet with carnitine and riboflavin supplements should be given.
Differential diagnosis
General differential considerations for certain features include:
- macrocephaly: few other leukodystrophies present with macrocephaly; these include Alexander disease, Canavan disease, L-2-hydroxyglutaric aciduria and megalencephalic leukoencephalopathy with subcortical cysts
- causes of bilateral basal ganglia high T2 intensity: amino acidopathies, mitochondrial diseases, Wilson's disease, Zellweger syndrome
- benign expansion of sub-arachnoid spaces (BESS): although prominent convexity spaces are common features, BESS is not associated with prominent CSF spaces in the sylvian fissures or anterior temporal poles. No parenchymal changes are seen either.
- non-accidental injury (NAI): glutaric aciduria type 1 is an important differential for NAI, as it can present with subdural hematomas
See also
- Alexander disease
- Canavan disease
- L-2-hydroxyglutaric aciduria
- megalencephalic leukoencephalopathy with subcortical cysts
- benign expansion of sub-arachnoid spaces
- non-accidental injury
Siehe auch:
- Morbus Wilson
- Battered-Child-Syndrom
- Morbus Alexander
- Morbus Canavan
- Zellweger-Syndrom
- 2-Hydroxy-Glutarazidurie
- Glutarylazidurie Typ 2
- Glutarazidurie
- megalencephalic leukoencephalopathy with subcortical cysts
- benign expansion of sub-arachnoid spaces
und weiter:


 Assoziationen und Differentialdiagnosen zu Glutarylazidurie Typ 1:
Assoziationen und Differentialdiagnosen zu Glutarylazidurie Typ 1: