RB




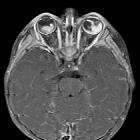
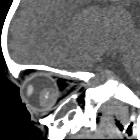





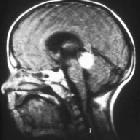

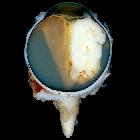

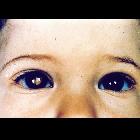






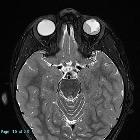
Retinoblastomas are the most common intraocular neoplasm found in childhood and with modern treatment modalities, are, in most cases, curable.
On imaging, they are generally characterized by a heterogeneous retinal mass with calcifications, necrotic components and increased vascularization on Doppler ultrasound/enhancement on CT/MRI.
Epidemiology
Retinoblastomas may be sporadic or secondary to a germline mutation of the retinoblastoma protein tumor suppressor gene (RB), which is usually inherited. It may be unilateral or bilateral:
- bilateral (30-40% of cases) essentially always have a germline mutation
- unilateral tumors (60-70% of cases) are caused by a germline mutation in approximately 15% of cases, whereas 85% are sporadic
Thus, ~55% of cases are due to a germline mutation. This mutation is inherited in an autosomal dominant fashion with ~90% penetrance (i.e. the child of a retinoblastoma survivor who has a germline mutation has a 50% chance of inheriting a mutation, and if they do so a 90% chance of developing a retinoblastoma. They thus have an overall probability of 45% of having a retinoblastoma (50% × 90%).
Most cases are diagnosed within the first four years of life, with a median age of diagnosis of 18-24 months .
Children with germline mutations are also at increased risk of developing trilateral retinoblastoma (unilateral or bilateral retinoblastomas and pineoblastoma) and osteosarcoma and usually present early (median age of diagnosis 12 months) .
Clinical presentation
Presentation is most frequently with leukocoria or loss of red-eye reflex. Overall approximately 30-40% are bilateral and often synchronous. The bilateral occurrence is even higher in inherited forms, and tend to occur at a younger age .
Pathology
Three patterns of growth are recognized :
- growth occurs inwards into the vitreous
- cell clusters may detach and float in the vitreous (vitreous seeding)
- growth occurs outwards
- associated with non-rhegmatogenous retinal detachment
Retinoblastoma may metastasize via direct spread into the orbit, along the optic nerve into the brain, or into the subarachnoid space resulting in leptomeningeal metastases. It can also haematogenously metastasize preferentially to the bone, bone marrow and liver. Rarely, it will spread to regional lymph nodes .
Macroscopic appearance
Macroscopic and funduscopic examination reveals a white elevated mass with fine surface vessels .
Histology
Histology demonstrates a small round-cell tumor of neuroepithelial origin. Flexner-Wintersteiner rosettes (relatively specific for retinoblastoma) and Homer-Wright pseudorosettes (also found in other PNETs) may be seen on microscopy.
Radiographic features
Ultrasound
Orbital sonography can be performed without sedation and can be repeated multiple times without exposing the child to ionizing radiation. Retinoblastomas appear as echogenic soft-tissue masses with variable shadowing due to calcifications and heterogeneity due to necrosis and hemorrhage . At diagnosis, tumors are usually vascular on Doppler examination.
The vitreous may have multiple areas of 'floating' debris, which may represent vitreous seeding or alternatively, necrotic debris, hemorrhage or increased globulin content .
CT
CT demonstrates a contrast-enhancing retrolental mass that is usually calcified. A dense vitreous due to hemorrhage is also common.
MRI
MRI is the modality of choice for pre-treatment staging on retinoblastoma (see retinoblastoma staging) .
- T1: intermediate signal intensity, hyperintense compared to the vitreous
- T2: hypointense compared to the vitreous
- T1 C+ (Gd)
- the mass usually enhances relatively homogeneously when small
- larger tumors often have areas of necrosis, rendering it heterogeneous
- linear enhancement of the choroid beyond the margins of the tumor should raise the possibility of choroidal involvement, although inflammation may lead to similar appearance
- enhancement of the anterior chamber need not represent tumor involvement, with hyperemia, uveitis, and iris neovascularization all leading to asymmetric enhancement
- careful assessment of the optic disc and optic nerve should be carried out to assess for involvement
- extra-ocular extension through the sclera will be visible as an interruption of the otherwise hypointense non-enhancing sclera by enhancing tumor
- DWI
Treatment and prognosis
Treatment depends on tumor size and the stage of disease (see retinoblastoma staging) and involves one or more modalities:
- conservative
- external-beam radiation therapy
- cryotherapy
- laser photocoagulation
- radioactive plaque therapy
- thermochemotherapy
- tumor reduction chemotherapy
- surgical
- enucleation
- en bloc resection
Prognosis depends on the stage. Overall the cure rate has risen to over 90% in first-world nations .
Patients with retinoblastoma rarely (<1%) go on to develop rhabdomyosarcoma, which itself perhaps arises due to chemotherapy, radiation therapy and/or an underlying genetic susceptibility .
Differential diagnosis
For imaging differentials consider:
- differential for calcification of the globe
- differential for an ocular mass
- differential for ocular enlargement
Siehe auch:
- Bulbus-oculi-Verkalkungen
- Aderhautmelanom
- Pineoblastom
- Tumoren des Auges
- trilateral retinoblastoma
- Morbus Coats
- retinoblastoma staging
- bilateral retinoblastoma
und weiter:
- intraorbitale Raumforderungen
- Drusen der Papilla nervi optici
- kapilläres Hämangiom der Orbita
- Leukokorie
- intraokulare Raumforderungen
- Pathologien des Auges
- Osteom der Aderhaut
- small round blue cell tumours
- malignancies in childhood
- Retinopathia praematurorum
- Retinale Dysplasie
- Phthisis bulbi oculi
- verdickter Nervus Opticus
- paediatric bone tumours (differential diagnosis)
- persistierender hyperplastischer primärer Glaskörper (PHPV)
- retinoblastoma: sonographic findings
- staging of retinoblastoma


 Assoziationen und Differentialdiagnosen zu Retinoblastom:
Assoziationen und Differentialdiagnosen zu Retinoblastom: