NMOSD















Neuromyelis optica (NMO) and neuromyelitis optica spectrum disorder (NMOSD) are closely related severe demyelinating diseases caused by an autoantibody to the aquaporin-4 water channel. The classic presentation of NMO is with the triad of optic neuritis, longitudinally extensive myelitis, and positive anti-AQP4 antibody, although a far wider range of manifestations are now recognized as part of NMOSD .
Terminology
Neuromyelitis optica (NMO) was previously referred to as Devic disease, and traditionally NMO was thought to have limited if any intracranial manifestations. Over the past decade, however, a far wider range of manifestations have been recognized as belonging together and thus the term NMOSD has been proposed to encompass them all.
Epidemiology
Neuromyelitis optica is typically found in patients somewhat older than those with multiple sclerosis (MS), with an average of presentation of 41 years, and there is an even stronger female predilection (F:M 6.5:1) . It is found more frequently in patients of Asian, Indian, and African descent .
Clinical presentation
NMO is characterized by bilateral optic neuritis and myelitis resulting in blindness and paraplegia. Although the two usually present concurrently, it is not uncommon for one to precede the other by up to several weeks . Additionally, it is now recognized that some patients present with unilateral optic nerve involvement.
Although NMO was initially thought of as a monophasic illness, it is now evident that, as with MS, it is usually a relapsing-remitting disease with symptomatic events separated by many years .
Furthermore, NMOSD also encompasses non-neurological manifestations in anti-AQP4 antibody seropositive patients including systemic lupus erythematosus (SLE) and Sjögren syndrome .
The current diagnostic criteria are defined by the International Panel for NMO Diagnosis.
Pathology
In approximately 70% (sensitivity of 70-90%; specificity of 90%) of patients with established NMO, a specific immunoglobulin can be isolated (AQP4-IgG which targets a transmembrane water channel (aquaporin 4) present on astrocyte foot processes abutting the limiting membrane . This accounts for some of the predilection for the circumventricular organs (e.g. periaqueductal grey matter) which are particularly rich in aquaporin 4 .
Early in the disease, demyelinating regions will demonstrate similar findings to MS, such as macrophage/microglia activation and axonal damage. Additionally, however, and relatively specific for NMO, these regions will also demonstrate extensive eosinophilic infiltration, perivascular deposition of immunoglobulins (especially IgM) and local activation of the complement cascade . Another differentiating feature is that axonal damage precedes demyelination in NMO .
Generally, the condition is sporadic, although some overlap in immunogenic features between certain viruses and aquaporin-4 water channel have been identified .
A significant proportion of individuals presenting with NMO but without AQP4 antibodies will be found to have anti-MOG IgG antibodies . These individuals appear to have somewhat different demographics and less severe clinical course .
Radiographic features
MRI
MRI is the modality of choice for NMOSD and the orbits, brain and spinal cord should be imaged in suspected cases.
Orbits
Targeted imaging of the orbits (including fat-saturated T1 C+ and T2 weighted sequences (see MRI protocol: orbit) may demonstrate typical features of optic neuritis:
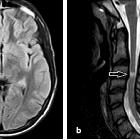
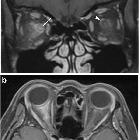
- optic nerves appearing hyperintense and swollen on T2 weighted sequences and enhancing on T1 C+
- bilateral optic nerve involvement and extension of the abnormal signal posteriorly as far as the chiasm is particularly suggestive of NMO
- atrophy of the optic nerves with associated hyperintensities on T2 weighted sequences may be seen in chronic stages of the disease
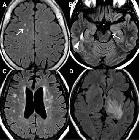
Brain
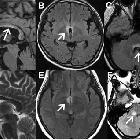
Although traditionally NMO was thought to have normal intracranial appearance it is increasingly evident that asymptomatic abnormalities are present in the majority of seropositive NMOSD patients. These can be divided into four categories :
- periventricular (hemispheric) confluent smooth sessile white matter involvement (unlike MS, there are usually no Dawson's fingers)
- periaqueductal grey matter
- hypothalamus/medial thalamus
- dorsal pons/medulla
- corpus callosum
- multiple callosal lesions with heterogeneous signal leading to a marbled pattern
- the splenium may be diffusely involved and expanded
- radially oriented (spilled-ink appearances) or spindle-shaped
- limited if any mass effect although tumefactive lesions do occur
- may involve overlying cortex
- facilitated diffusion
- may be more common in children and in patients from the Far East and Africa
- often vanish but cystic change is seen in a minority of cases
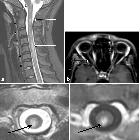
Spinal cord
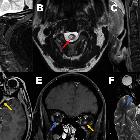
Spinal cord involvement is extensive, with high T2 signal spanning at least three vertebral segments, often many more (known as a longitudinally extensive spinal cord lesion) . Cord swelling is usually present in the acute phase. Although less common, short transverse myelitis is seen in 14.5% of cases .
Bright spotty lesions are a specific feature of NMO. It consists of marked T2 hyperintense (higher than CSF) and T1 hypointense foci in the central grey matter.
Imaging features include :
- T1
- hypointense
- follow-up scans may demonstrate cord atrophy and low T1 signal
- T2
- hyperintense (often >3 vertebral body lengths)
- central grey matter involvement
- bright spotty lesions (see above)
- T1 C+ (Gd)
- enhancement is common and variable in appearance
- ring-enhancement
- seen in a third of patients
- both on sagittal and axial imaging
- often ring extends over multiple vertebral levels
- patchy "cloud-like" enhancement of the aforementioned T2 bright lesions may be present
- thin ependymal enhancement similar to ependymitis
- lens-shaped enhancement on sagittal images
Treatment and prognosis
Treatment of NMO is evolving, with immunosuppression (e.g. anti-CD20 monoclonal antibody rituximab) appearing effective .
It is important to distinguish NMO from MS as the treatment not only is different but treating a patient with NMO with MS-specific therapies (e.g. beta-interferon or natalizumab) can actually lead to its exacerbation .
Patients with a relapsing course have a poorer prognosis :
- blind in one or both eyes: monophasic 22% vs. relapsing 60%
- monoplegia or paraplegia: monophasic 31% vs. relapsing 52%
History and etymology
The disease was first described in 1870 by Sir Thomas Clifford Allbutt . The French term "neuromyélite optique aiguë" (literally acute optic neuromyelitis) was later coined by Dr. Eugène Devic and Fernand Gault in 1894 in a case series of 16 patients . At the time, the disorder was thought to be acute and monophasic.
For many years, it was considered to be a subtype of multiple sclerosis, until the discovery of anti-aquaporin-4 antibodies at which time it was moved into its own disease category. It is now evident that a significant proportion of patients with clinical NMO do not have the anti-AQP4-antibody (some have anti-MOG-antibodies for example) and that the presentation can be more heterogeneous. At present, therefore, the term neuromyelitis optica spectrum disorder (NMOSD) is now used. It is very likely that there will be significant further evolution of our understanding of this disease in the near future.
Differential diagnosis
The differential diagnosis depends on the presentation, and when classic, the diagnosis can be made with a fair degree of certainty. The most important clinical and imaging differential is MS as both can present with optic neuritis, cerebral and spinal demyelination including involvement of the corpus callosum (see Practical points).
For patients with cerebral involvement, the differential is broad and includes:
- multiple sclerosis
- Susac syndrome: involves the central portion of the corpus callosum
- neuro-Behçet: mesodiencephalic involvement is typical
- primary angiitis of the CNS (PACNS)
- acute disseminated encephalomyelitis (ADEM): grey-white matter involvement with a more tumefactive appearance
- amyotrophic lateral sclerosis (ALS): bilateral corticospinal tract involvement is more symmetrical
For patients with optic nerve involvement, the differential is that of optic neuritis.
For patients with spinal cord involvement, the differential is that of a longitudinally extensive spinal cord lesion. For the typical bright spotty lesion, an important differential is acute spinal cord ischemia.
Practical points
There are many features that distinguish NMO from multiple sclerosis, although none are pathognomonic. It should also be noted that overlap of imaging findings of these two entities may be higher in Asian populations . Nonetheless, features that are helpful in favoring NMO over multiple sclerosis include :
- brain
- smooth confluent periependymal distribution (see above)
- fewer oval perivenular orientation of periventricular lesions (no Dawson's fingers)
- fewer juxtacortic lesions (U-fiber)
- more extensive involvement of the corpus callosum (especially its ependymal surface)
- larger, more confluent lesions
- lack of open ring enhancement
- corticospinal tract and diencephalic involvement (see above)
- spinal cord
- more longitudinally extensive spinal cord lesion
- preferential involvement of the central cord rather than the peripheral cord
- more longitudinally extensive spinal cord lesion
- optic nerves
- more longitudinally extensive optic neuritis
- with preferential involvement of the posterior optic pathway
- more longitudinally extensive optic neuritis


 Assoziationen und Differentialdiagnosen zu Neuromyelitis optica:
Assoziationen und Differentialdiagnosen zu Neuromyelitis optica: