Encephalomyelitis disseminata
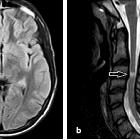











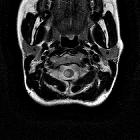















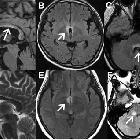




Multiple sclerosis (MS) is a relatively common acquired chronic relapsing demyelinating disease involving the central nervous system, and is the second most common cause of neurological impairment in young adults, after trauma . Characteristically, and by definition, multiple sclerosis is disseminated not only in space (i.e. multiple lesions in different regions of the brain) but also in time (i.e. lesions occur at different times).
A number of clinical variants are recognized, each with specific imaging findings and clinical presentation. They include:
- classic multiple sclerosis (Charcot type)
- tumefactive multiple sclerosis
- Marburg type (acute malignant)
- Schilder type (diffuse cerebral sclerosis)
- Balo concentric sclerosis
This article concerns itself primarily with classic (Charcot type) multiple sclerosis. The other variants are discussed separately.
Importantly, neuromyelitis optica (Devic disease) was considered a variant of multiple sclerosis, but is now recognized as a distinct entity, and is therefore also discussed separately.
Epidemiology
The presentation is usually between adolescence and the sixth decade, with a peak at approximately 35 years of age . There is a strong, well recognized female predilection with a F:M ratio of approximately 2:1 .
Multiple sclerosis has a fascinating geographic distribution: it is rarely found in equatorial regions (e.g. 15 per 100,000), with incidence gradually increasing with distance from the equator (e.g. 250 per 100,000) .
Clinical presentation
Clinical presentation is both highly variable acutely, as a result of varying plaque location, as well as over time. Examples of common clinical features include :
- brainstem and cranial nerve involvement:
- optic neuritis
- internuclear ophthalmoplegia (often bilateral)
- trigeminal neuralgia
- diplopia (e.g. due to abducens nerve palsy)
- vertigo
- cerebellum involvement:
- ataxia and gait disturbance
- oscillopsia
- cerebrum and spinal cord involvement:
- limb sensory loss or paresthesias
- upper motor neuron signs
- Lhermitte sign
- urinary incontinence
- others:
- fatigue
- depression
- Uhthoff phenomenon: heat and exercise worsen symptoms
- cognitive decline
Classification
A number of patterns of longitudinal disease have been described :
- most common (70% of cases)
- patients exhibit periodic symptoms with complete recovery (early on)
- approximately 85% of patients with relapsing-remitting MS eventually enter a secondary progressive phase
- uncommon (10% of cases)
- patients do not have remissions, with neurological deterioration being relentless
- 15-50% of cases
- defined as patients who remain functionally active for over 15 years
As is evident from this list, there is overlap, and in some cases, patients can drift from one pattern to another.
Upon presentation patients often have evidence of multiple previous asymptomatic lesions, and the diagnosis of multiple sclerosis can be strongly inferred. In other instances patients present with the first plaque. This is known as clinically isolated syndrome (CIS) and not all patients go on to develop multiple sclerosis.
Radiologically isolated syndrome (RIS) is another entity based on MRI brain findings which described as incidental white matter lesions suggestive of MS on imaging in a patient without associated clinical symptoms .
Diagnosis
The diagnosis of multiple sclerosis requires the constellation of clinical findings and various investigations (see McDonald diagnostic criteria for multiple sclerosis), including :
- typical history
- oligoclonal bands in CSF
- immunoglobulin G in serum
- abnormal visual evoked potential
- MR imaging
- lack of viable alternative diagnosis
Pathology
The exact etiology is poorly known although it is believed to have both genetic and acquired contributory components. An infectious agent (e.g. EBV), or at least a catalyst, has long been suspected due to the geographic distribution and presence of clusters of cases; however, no agent has yet been firmly confirmed. Some authors also suggested that "chronic cerebrospinal venous insufficiency" can cause or exacerbate MS but this theory has not been proven by further investigations .
Multiple sclerosis is believed to result from a cell-mediated autoimmune response against one's own myelin components, with loss of oligodendrocytes, with little or no axonal degeneration in the acute phase; however, in later stages, loss of oligodendrocytes results in axonal degeneration.
Demyelination occurs in discrete perivenular foci, termed plaques, which range in size from a few millimeters to a few centimeters .
Each lesion goes through three pathological stages:
- early acute stage (active plaques)
- active myelin breakdown
- plaques appear pink and swollen
- subacute stage
- plaques become paler in color ("chalky")
- abundant macrophages
- chronic stage (inactive plaques/gliosis)
- little or no myelin breakdown
- gliosis with associated volume loss
- appear grey/translucent
Associations
- a strong association with HLA-DR2 class II has been identified
- Melkersson-Rosenthal syndrome: postulated
Radiographic features
Plaques can occur anywhere in the central nervous system. They are typically ovoid in shape and perivenular in distribution.
CT
CT features are usually non-specific, and significant change may be seen on MRI with an essentially normal CT scan. Features that may be present include:
- plaques can be homogeneously hypoattenuating
- brain atrophy may be evident in with long-standing chronic MS
- some plaques may show contrast enhancement in the active phase
MRI
MRI has revolutionised the diagnosis and surveillance of patients with MS. Not only can an MRI confirm the diagnosis (see McDonald diagnostic criteria for multiple sclerosis), but follow-up scans can assess response to treatment and help determine the disease pattern.
Protocol
Although many sequences are contributory, the 2018 Revised Guidelines of the Consortium of MS Centers MRI Protocol for the Diagnosis and Follow-up of MS plaques lists the following core sequences :
- FLAIR (axial and sagittal)
- ideally performed as a 3D volumetric scan (1 mm isotropic), or
- 3 mm contiguous
- T1: 3D inversion recovery prepared gradient echo
- T2 (axial): 3D or 2D
- DWI (axial)
Note: contrast is not necessary for routine asymptomatic follow-up.
Sequence utility
- T1
- lesions are typically iso- to hypointense (T1 black holes)
- callososeptal interface may have multiple small hypointense lesions (Venus necklace) or the corpus callosum may merely appear thinned
- hyperintense lesions are associated with brain atrophy and advancing disease
- T2
- lesions are typically hyperintense
- acute lesions often have surrounding edema
- SWI
- central vein sign: at higher field strengths most plaques have been shown to be perivenular (at 3 T, 45% of lesions; at 7 T, 87% of lesions)
- FLAIR
- lesions are typically hyperintense
- a very early sign is called ependymal dot-dash sign
- when these propagate centrifugally along the medullary venules and arranged perpendicular to the lateral ventricles in a triangular configuration (extending radially outward - best seen on parasagittal images), they are termed Dawson's fingers
- FLAIR is more sensitive than T2 in the detection of juxtacortical and periventricular plaques, while T2 is more sensitive to infratentorial lesions
- T1 C+ (Gd)
- active lesions show enhancement
- enhancement is often incomplete around the periphery (open ring sign)
- DWI/ADC
- active plaques may demonstrate high or low ADC (increased or decreased diffusion)
- also typically open ring in morphology
- Proton density(PD)
- proton density images are better at detecting cervical spinal cord MS lesions especially when T2W images fail to demonstrate these lesions
- MR spectroscopy
- NAA peaks may be reduced within plaques, which is the most common and remarkable finding
- choline and lactate are found to be increased in the acute pathologic phase
- double inversion recovery DIR
- a new sequence that suppresses both CSF and white matter signal and offers better delineation of the plaques
Location of the plaques can be infratentorial, in the deep white matter, periventricular, juxtacortical or mixed white matter-grey matter lesions.
Even on a single scan, some features are helpful in predicting relapsing-remitting vs. progressive disease. Features favoring progressive disease include:
- large numerous plaques
- hyperintense T1 lesions
Treatment and prognosis
The aim of treatment is twofold: to curtail progression (disease-modifying agents) and symptomatic relief.
Steroids, interferon, monoclonal antibodies and autologous hematopoietic stem cell transplantation are all used. Although discussion of individual agents and therapies is well beyond the scope of this article, it is worth being aware of the main agents available and their mechanism of action :
- interferon beta: inhibition of T-lymphocyte proliferation
- glatiramer acetate: immunomodulation
- teriflunomide (Aubagio): reduces both T-cell and B-cell activation and proliferation
- dimethyl fumarate (Tecfidera): immunomodulation
- fingolimod (Gilenya): prevents lymphocyte migration out of lymph nodes and into CNS
- natalizumab (Tysabri): inhibits binding of lymphocytes to endothelium
- alemtuzumab (Lemtrada): immunomodulation of T-cell and B-cell function
- mitoxantrone: reduces T-cell and B-cell proliferation and reduces T-cell activation
Complications
In addition to the potential for disease progression resulting in progressive neurological impairment, a number of specific complications need to be considered. These include :
- progressive multifocal leukoencephalopathy (PML) and other less common CNS JC virus manifestations (JC virus granule cell neuronopathy, JC virus encephalopathy, and JC virus meningitis)
- particularly in patients treated with natalizumab with positive JC virus serology
- PML-IRIS
- a complication of cessation of natalizumab or treatment for natalizumab-related PML with plasma exchange or immunoabsorption
- primary CNS lymphoma
- rarely lymphoma appears to arise from previously identified demyelinating lesions
Prognosis
Prognosis is variable and depends on the pattern of disease a patient has (e.g. primary progressive carries a worse prognosis than relapsing-remitting).
In general, patients with relapsing-remitting MS will progress to secondary progressive disease in 10 years and will require ambulatory aids (e.g. cane/wheelchair/frame) in another 5 to 15 years . Approximately half of the affected individuals will no longer be independently ambulatory after 20 years .
Overall life expectancy is also reduced, by 7 to 14 years .
Differential diagnosis
The differential diagnosis is dependent on the location and appearance of demyelination. For classic (Charcot type) MS, the differential can be divided into intracranial and spinal involvement.
For intracranial disease, the differential includes almost all other demyelinating diseases as well as:
- CNS fungal infection (e.g. Cryptococcus neoformans) - patients tend to be immunocompromised
- mucopolysaccharidosis (e.g. Hurler disease) - congenital and occurs in a younger age group
- Marchiafava-Bignami disease (for callosal lesions)
- Susac syndrome
- CNS manifestations of primary antiphospholipid syndrome
For spinal involvement, the following should be considered:
- transverse myelitis
- infection
- spinal cord tumors (e.g. astrocytomas)
Multiple sclerosis variants (e.g. tumefactive MS, Devic disease) are discussed separately.


 Assoziationen und Differentialdiagnosen zu Encephalomyelitis disseminata:
Assoziationen und Differentialdiagnosen zu Encephalomyelitis disseminata: