progressive multifocal leukoencephalopathy














Progressive multifocal leukoencephalopathy (PML) is a demyelinating disease which results from the reactivation of John Cunningham virus (JC virus) infecting oligodendrocytes in patients with compromised immune systems. It is considered the most common clinical manifestation of John Cunningham virus (JC) virus infection in the brain , and is seen in three clinical contexts:
Epidemiology
Progressive multifocal leukoencephalopathy is strongly associated with immunosuppressed states, and primary PML developing in an immunocompetent patient is very rare.
Classically, PML occurred in patients with AIDS, typically developing in patients with CD4 counts of 50-100 cells/μL, and is found in approximately 5% of autopsies of patients who died from AIDS .
However, the incidence of PML in the non-HIV setting is thought to be increasing :
- post-transplant: bone marrow or solid organ transplants
- leukemia
- solid organ malignancies
- inflammatory diseases (e.g. SLE, sarcoidosis)
- isolated CD4 lymphocytopaenia
- immunosuppressive monoclonal antibody therapy, such as natalizumab (an IgG monoclonal antibody used in the treatment of relapsing-remitting multiple sclerosis ), efalizumab, and rituximab
In addition to being seen in immunocompromised patients, it is also seen in patients whose immune function is recovering, and PML is one of the classic conditions encountered in immune reconstitution inflammatory syndrome (IRIS) .
Clinical presentation
Patients with progressive multifocal leukoencephalopathy present with various neurological symptoms. It typically spares the optic nerve and the spinal cord. The most frequently encountered symptoms include:
- altered mental status
- motor deficits
- limb and gait ataxia
- visual symptoms (diplopia and hemianopia)
- seizure (as PML can also involve the grey matter in later stages)
The final diagnosis is established with brain biopsy (specificity: 100%, sensitivity: 65-95%).
Pathology
Etiology
PML lesions are secondary to progressive demyelination due to John Cunningham (JC) virus (usually reactivation) which infects oligodendrocytes. In PML seen in HIV/AIDS, reactivation is typically seen when CD4 cell count drops below 100/μL .
Location
Lesions tend to have a confluent, bilateral but asymmetrical supratentorial white matter and thalamic involvement . However, basal ganglia, brainstem and cerebellum also can be involved. Subcortical frontal and parieto-occipital regions are common locations. However, isolated posterior fossa involvement has also been reported . While the condition invariably involves white matter, subcortical U-fibers and in late stages of disease grey matter involvement is also seen .
Microscopic appearance
Histology reveals demyelinating plaques involving the white matter and subcortical U-fibers. Other findings include infected oligodendrocytes with enlarged amphophilic nuclei located at the periphery of the lesions, macrophages containing phagocytosed cellular debris and myelin, and reactive gliosis with enlarged astrocytes .
Radiographic features
CT
Asymmetric focal zones of low attenuation involving the periventricular and subcortical white matter. This is in contradistinction to more symmetrical hypoattenuation seen in HIV encephalopathy.
MRI
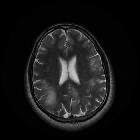
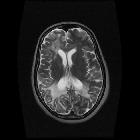
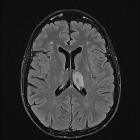
Typically seen as multifocal, asymmetric periventricular and subcortical involvement. There is little, or no mass effect or enhancement and the subcortical U-fibers are commonly involved with a predilection for the parieto-occipital regions . In fact, even though subcortical U-fiber involvement is an important feature of lesions of multiple sclerosis, these are relatively uncommon and if new lesions have a subcortical U-fiber distribution, this favors PML . Typically, the lesion presents a sharply demarcated peripheral border along the subcortical U fibers and a hazy and ill-defined inner border. Corpus callosum may be involved .
- T1: involved regions are usually hypointense
- T2:
- involved regions are hyperintense
- multiple punctate high T2 signal lesions surrounding the main area (milky way sign)
- barbell sign: parieto-occipital signal abnormality crossing the splenium
- T1 C+ (Gd)
- typically there is no enhancement
- enhancement can be seen in PML-IRIS, AIDS with HAART, and in patients on natalizumab
- when enhancement is present, it may be associated with improved survival
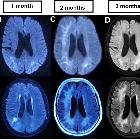
- ADC/DWI: peripheral patchy diffusion restriction particularly at the leading edge
- MR spectroscopy: according to one study, spectra of PML lesions were characterized by significantly reduced NAA, lactate presence, and by significantly increased choline and lipids compared with control group values
- MR perfusion:
- elevated at the leading edge of lesions
- most commonly seen in progressive non-IRIS PML
Treatment and prognosis
Prognosis is generally poor with an inexorable neurological decline leading to comaand death occurring in the majority of patients with PML . If untreated, PML is usually fatal within one year, often within 2 to 6 months . Treatment with highly active antiretroviral therapy (HAART) may prolong survival, although in some instances this leads to PML-IRIS. High-dose glucocorticoid therapy is recommended for patients with PML-IRIS. Some reports also state some benefit with cytarabine or mirtazapine, the latter especially in natalizumab-associated PML.
Differential diagnosis
- new demyelinating lesions in patients with multiple sclerosis
- early in the disease MS and PML may appear similar
- periventricular location or well-defined borders favor new MS lesions
- HIV/AIDS encephalopathy: often diffuse white matter disease, with atrophy, symmetric, spares the subcortical U-fibers
- posterior reversible encephalopathy syndrome (PRES)
- differing clinical history (e.g. hypertension)
- can involve both grey and white matter
- acute disseminated encephalomyelitis (ADEM)
- differing clinical history (e.g. recent infection/vaccination)
- can involve both grey and white matter
- lesions usually enhance
- cerebral toxoplasmosis: usually enhance
- Immunodeficiency-associated CNS lymphomas: usually enhance
See also
Siehe auch:
- posteriores reversibles Enzephalopathiesyndrom
- Demyelinisierende Erkrankung
- Akute disseminierte Enzephalomyelitis
- CNS manifestations of AIDS
- Variables Immundefektsyndrom
- relapsing remitting multiple sclerosis
- AIDS
und weiter:


 Assoziationen und Differentialdiagnosen zu Progressive multifokale Leukenzephalopathie:
Assoziationen und Differentialdiagnosen zu Progressive multifokale Leukenzephalopathie:


