sarcoidosis








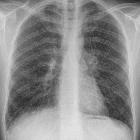
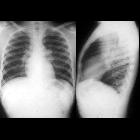
























Sarcoidosis is a non-caseating granulomatous multisystem disease with a wide range of clinical and radiographic manifestations.
Individual systemic manifestations are discussed in respective articles:
- pulmonary and mediastinal manifestations
- cardiac manifestations
- musculoskeletal manifestations
- head and neck manifestations
- central nervous system manifestations
- abdominal manifestations
- cutaneous manifestations
The remainder of this article pertains to a general discussion of sarcoidosis.
Epidemiology
Sarcoidosis occurs all over the world in all ages and races. Attempts to describe accurate epidemiology are complicated by the use of inconsistent diagnostic criteria and variable (often asymptomatic) disease manifestations.
Still, certain epidemiological patterns are reported in the literature:
- age of onset
- most commonly presents between 2 through 4 decades of life, although diagnosis in children and elderly also recognized
- gender
- inconsistent data
- there may be a small female predominance among African-Americans
- race
- highest incidence in African-Americans (36 to 50 per 100,000) and northern European Caucasians
- incidence among Caucasians has been estimated at 11 to 20 per 100,000
- reported incidence is lower in Asian populations
- heritability
- familial clustering of sarcoidosis has been reported, suggesting either a genetic or environmental component of the disease
- the two largest studies suggest a familial relative risk increase ~4x given a single affected first-degree relative
Clinical presentation
Clinical presentation is variable and diagnosis is usually made on the combination of clinical and radiological features.
- half of the patients are asymptomatic
- the rest develop respiratory (e.g. cough and dyspnea) or skin (e.g. erythema nodosum, lupus pernio, scars, plaques) disease
- ocular, lacrimal gland and salivary gland involvement are relatively common
- approximately 5% of patients develop neurosarcoidosis
Additionally, patients may have specific syndromic presentations:
- Löfgren syndrome: an uncommon but specific acute presentation of sarcoidosis with a better prognosis
- Heerfordt syndrome: an uncommon variant of sarcoidosis comprising of fever, parotid enlargement, facial palsy, and anterior uveitis
Pathology
Sarcoidosis is a multisystem disorder of unknown etiology characterized by the formation of inflammatory non-caseating granulomas within affected tissues. Histologically, the lesions characteristically demonstrate an absence of a necrotic component, except in rare cases (so-called "necrotizing sarcoid granulomatosis"). The granulomas may resolve spontaneously or progress to fibrosis.
It is thought to represent a disorder of immune regulation, particularly of cell-mediated immunity . Mycobacterium and propionibacterium RNA and DNA have been detected in sarcoidosis lesions, raising the possibility of an infectious trigger . Reported familial clustering and co-occurrence in monozygotic twins suggest that genetic susceptibility may also play an important role.
Microscopic appearance
Characteristic (but not pathognomonic) histological features include:
- non-caseating granulomas
- epithelioid cells, macrophages, multinucleated giant cells, and CD4+ T cells
- fibroblasts, B lymphocytes, and CD8+ T cells may be present more peripherally
- Schaumann bodies: laminated calcium-containing concretions
- asteroid bodies
- necrosis is atypical and may suggest another etiology
Markers
Biochemical markers include:
- elevated angiotensin-converting enzyme (ACE)
- 40% false negative
- 10% false positive
- hypercalcemia and hypercalciuria
Additionally, a positive Kveim-Siltzbach skin test helps to confirm the diagnosis .
Radiographic features
90% of patients have pulmonary involvement (although many are asymptomatic) . Since chest x-rays are readily available and have low radiation burden, the pattern of nodal and parenchymal involvement is typically used to 'stage' sarcoidosis (chest x-ray staging of sarcoidosis) .
Treatment and prognosis
Treatment is primarily with judicious use of corticosteroids, to avoid adverse medication effects.
Pulmonary involvement is responsible for the majority of morbidity and mortality in patients with sarcoidosis. The overall mortality rate is approximately 5%, with patients who present insidiously faring worse than those who present with an acute onset . Likelihood of resolution depends on the stage of disease at presentation :
- stage I: 60% resolution within 1-2 years
- stage II: 46%
- stage III: 12%
Complications
Complications depend on the systemic pattern of disease involvement. As pulmonary sarcoidosis is by far the most common manifestation, so are thoracic complications. They include :
- pulmonary fibrosis (stage IV)
- pulmonary arterial hypertension and cor pulmonale
- aspergillomas, which may be complicated by hemoptysis
History and etymology
Historically, sarcoidosis was also known as Besnier-Boeck-Schaumann disease/syndrome, or various combinations thereof, e.g. Besnier-Boeck disease, Boeck disease, or Schaumann syndrome .
Sarcoidosis is derived from the Ancient Greek terms, σαρξ (sarc) meaning flesh, ειδος (eidos) meaning form, and σις (-sis), a suffix meaning a process .
Siehe auch:
- Sarkoidose der Milz
- Sarkoidose der Leber
- Neurosarkoidose
- Lungenfibrose
- Aspergillom
- pulmonale und mediastinale Sarcoidose
- pulmonale Hypertonie
- bihiläre Lymphadenopathie
- Sarkoidose Stadien
- löfgren syndrome
- Garland Trias
- abdominelle Manifestationen der Sarkoidose
- musculoskeletal manifestations of sarcoidosis
- heerfordt syndrome
- panda sign
- kardiale Beteiligung bei Sarkoidose
- Berylliose
- sarcoid galaxy sign
- lambda sign
und weiter:
- Lymphangiosis carcinomatosa
- verkalkte Lungenherde
- Nervus facialis
- Langerhanszell-Histiozytose der Lunge
- Kerley-Linien
- Silikose
- verkalkte mediastinale Lymphknoten
- Kryptogene organisierende Pneumonie (COP)
- Bronchiolitis obliterans
- right middle lobe consolidation
- Funikuläre Myelose
- Splenomegalie
- Granulomatose mit Polyangiitis
- hiläre Lymphadenopathie
- Ollier-Syndrom
- Warthin-Tumor
- animal and animal produce inspired signs
- akute Pyelonephritis
- Meningitis
- multiple zystische Lungenherde
- Chylothorax
- mediastinal lymphoma
- Dural-Tail-Zeichen
- Metastasen in der Orbita
- Sarkoidose ossäre Manifestationen
- pulmonary opacification
- miliare Lungenherde
- crazy paving-Muster
- Meningeom Nervus opticus
- Interstitielle Lungenerkrankung
- apical zone
- Erythema nodosum
- pulmonale venookklusive Erkrankung
- bilaterale hiläre Lymphadenopathie
- fibrosierende Mediastinitis
- differential of chronic alveolar opacities
- bilaterale axilläre Lymphadenopathie
- conditions involving skin and bone
- sausage digit
- hyperdenser Lymphknoten
- Parenchymband
- stroke in children and young adults
- Eierschalenverkalkungen
- Nasenseptumdefekt
- chronic bilateral airspace opacification
- hyperechoic liver
- testicular sarcoidosis
- pulmonale Calciphylaxie
- diffuse Trachealwandverdickung
- calcifying epithelioma of Malherbe
- echogenic renal pyramids
- hypervaskularisierte Lymphknoten
- obliterative bronchiolitis (mnemonic)
- generalised increased liver echogenicity
- adenoid cystic carcinoma of lacrimal glands
- idiopathische granulomatöse Mastitis
- diffuse Trachealverengung
- pulmonary arterial hypertension classification - third world symposium on PAH
- Läsionen der Fingerspitze
- verdickter Nervus Opticus
- Silzbach sarcoidosis chest radiographic staging system
- bronchovascular spread (mnemonic)
- lambda sign of sarcoidosis
- sarcoidosis: musculoskeletal manifestations of the skull
- Verkalkungen bei Sarkoidose
- vertebral sarcoidosis
- tuberkulöse Daktylitis
- Bronchusobstruktion durch granulomatöse Erkrankungen (Sarkoidose)
- Merkspruch Nasenseptumperforation
- Eierschalenverkalkungen von Lymphknoten
- benigne lymphoepitheliale Läsionen
- Sarkoidose der Hypophyse
- Kveim Stilzbach skin test
- Pulmonalvenenstenose
- chronische Periaortitis
- Metastasen in der Hypophyse


 Assoziationen und Differentialdiagnosen zu Sarkoidose:
Assoziationen und Differentialdiagnosen zu Sarkoidose: 









