eosinophilic granuloma of the lung























Pulmonary Langerhans cell histiocytosis (PLCH) may be seen as part of widespread involvement in patients with disseminated Langerhans cell histiocytosis or more frequently as a distinct entity in young adult smokers. This article focuses on the latter.
Epidemiology
Pulmonary Langerhans cell histiocytosis is usually identified in young adults (20-40 years of age). A history of current or previous cigarette smoking is identified in up to 95% of cases . It is a rare disorder with no well-established gender predilection, which appears to be more common in Caucasian populations .
Associations
Hematopoietic neoplasms:
Clinical presentation
Presentation is usually with dyspnea or non-productive cough. Other symptoms include constitutional symptoms (fatigue and weight loss), pleuritic chest pain, or spontaneous pneumothorax . Up to a quarter of patients are asymptomatic.
Pathology
Langerhans cells proliferate in the bronchiolar and bronchial epithelium, forming granulomas. It is postulated that as these cellular granulomas evolve, peripheral fibrosis forms resulting in traction on the central bronchiole which becomes cyst-like . This explains the presumed evolution from a nodule, through cavitating nodule and thick-walled cysts, to the 'stable' thin-walled cysts . An immune-mediated mechanism has been postulated, although an inciting agent has not been isolated . This proliferation is accompanied by inflammation and granuloma formation. Electron microscopy may reveal characteristic Birbeck granules .
More recent evidence suggests that pulmonary Langerhans cell histiocytosis represents a myeloid neoplasm with inflammatory properties .
Radiographic features
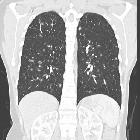
Pulmonary Langerhans cell histiocytosis has variable appearance depending on the stage of the disease, ranging from small peribronchiolar nodular opacities to multiple irregularly-shaped cysts. There is a mid and upper zone predilection .
Plain radiograph
The earliest change is a diffuse bilateral symmetrical reticulonodular pattern with a predilection for the mid and upper zones. The ill-defined nodules range from 1-10 mm in size. Later, cyst formation may be seen or may mimic a honeycomb appearance due to a summation of air-filled cysts. Cysts can be identified in only 1-15% of cases , and range from 1-3 cm in diameter. There is a preservation of lung volumes or even hyperinflation . Reduced lung volumes are uncommon and only seen in end-stage fibrotic cases . Lymph node enlargement visible on chest x-rays is rare .
CT
As is usually the case, CT and especially HRCT is superior to plain chest radiography in identifying both the reticulonodular opacities and cysts . Distribution is the key in differentiating pulmonary Langerhans cell histiocytosis from other cystic lung diseases with a predilection for the mid and upper zones and regional sparing of the costophrenic recesses, anterior right middle lobe and lingula left upper lobe .
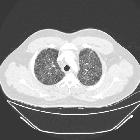
- nodules
- more pronounced early in the disease
- may range in number from a few to innumerable
- 1-10 mm in diameter (typically 1-5 mm )
- centrilobular distribution - may also be peribronchial or peribronchiolar
- usually have irregular margins
- may be cavitary nodules with thick walls, later becoming cysts
- surrounding lung parenchyma appears normal
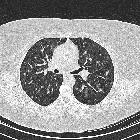
- cysts
- more pronounced later in the disease
- usually less than 10 mm in diameter
- may measure up to 2-3 centimeters in size
- the extreme bases may be preserved
- usually thin-walled, but on occasion may be up to a few millimeters thick
- confluence of 2 or more cysts results in bizarre shapes
- bilobed
- cloverleaf
- branching
- internal septations
Other common findings include :
- ground-glass and/or reticular opacities
- DIP-like change
- mosaic attenuation
- septal line thickening
- emphysema
In late disease, other findings include:
- coalescent cysts
- fibrosis
- honeycombing
The appearance of new nodules later in the disease (when cystic change is established) indicates disease progression but is a rare finding .
Treatment and prognosis
Overall prognosis is generally good with over 50% of patients demonstrating spontaneous resolution or stabilization even without treatment . This is especially the case in patients who stop smoking.
In a minority of patients (~20%) and more frequently in those who continue to smoke, the disease is progressive with deterioration in respiratory function and eventual end-stage pulmonary fibrosis .
Treatment may not be required once smoking has ceased. Corticosteroids are frequently used and appear beneficial. In patients with rapidly progressive disease, no proven therapy has been found. In some selected patients lung transplantation may be an option, provided smoking has ceased. Recurrence in the transplanted lung has been described .
Complications
- cyst rupture
- spontaneous pneumothorax: may be the first presentation
- pneumomediastinum
- interstitial fibrosis
- pulmonary arterial hypertension and cor pulmonale
- end-stage pulmonary fibrosis and respiratory failure
Differential diagnosis
Differential depends on whether the nodular or cystic change is the dominant feature.
Early in the disease, when nodules are the dominant feature, consider:
-
granulomatous disease
- metastases
- miliary tuberculosis
See differential of multiple pulmonary nodules and differential of miliary opacities for more comprehensive lists.
Later in the disease, when cysts are prominent, consider:
- lymphangiomyomatosis (LAM)
- diffuse distribution
- regular shaped and sized cysts
- cystic bronchiectasis from ABPA
- central distribution
- mucous plugging
- centrilobular emphysema
- lack of visible cyst wall
- Pneumocystis jiroveci pneumonia
- idiopathic pulmonary fibrosis
- basal and subpleural distribution
- reduced lung volumes
- lymphocytic interstitial pneumonitis (LIP)
- smooth-walled simple cysts
- associated with autoimmune disease
See also
- Langerhans cell histiocytosis: general discussion
- Letterer-Siwe disease
- Hand-Schüller-Christian disease
- eosinophilic granuloma (EG)
Siehe auch:
- Pneumothorax
- Sarkoidose
- mosaikartige Verdichtungen der Lunge
- Mediastinalemphysem
- Granulomatose mit Polyangiitis
- Lymphangioleiomyomatose
- Allergische bronchopulmonale Aspergillose
- Honigwabenmuster
- multiple zystische Lungenherde
- pulmonale Hypertonie
- Pneumocystis jiroveci Pneumonie
- Histiozytose X
- skeletale Manifestationen der Langerhanszell-Histiozytose
- Emphysem
- Desquamative interstitielle Pneumonie (DIP)
- zentrilobuläre Verteilung
- ground-glass opacities
- differential of miliary opacities
und weiter:


 Assoziationen und Differentialdiagnosen zu Langerhanszell-Histiozytose der Lunge:
Assoziationen und Differentialdiagnosen zu Langerhanszell-Histiozytose der Lunge:













