lymphangiomyomatosis



























Lymphangioleiomyomatosis (LAM) is a rare multi-system disorder that can occur either sporadically or in association with the tuberous sclerosis complex (TSC) and is often considered a forme fruste of TSC.
Epidemiology
It almost exclusively affects women of childbearing age . The estimated incidence is 1:400,000 .
Clinical presentation
Patients usually present with exertional dyspnea and recurrent episodes of pneumothorax are common .
In 2011, the European Respiratory Society published guidelines for the diagnosis and management of lymphangioleiomyomatosis, which has established the following diagnostic criteria :
- definite LAM
- characteristic or compatible lung HRCT and lung biopsy fitting the pathological criteria for LAM or
- characteristic lung HRCT and any of the following
- renal angiomyolipoma
- thoracic or abdominal chylous effusion
- lymphatic malformations or lymph-node involved by LAM
- definite or probable tuberous sclerosis
- probable LAM
- characteristic HRCT and compatible clinical history or
- compatible HRCT and any of following
- renal angiomyolipoma
- thoracic or abdominal chylous effusion
- possible LAM
- characteristic or compatible HRCT
In 2016, the American Thoracic Society and the Japanese Respiratory Society published clinical practice guidelines that state a definitive diagnosis of LAM can be established if the patient :
- has a compatible clinical history: young to middle-aged, female, presenting with worsening dyspnea and/or pneumothorax/chylothorax in the absence of features suggestive of other cystic lung diseases
- has a characteristic HRCT of the chest
- has one or more of the following features
- tuberous sclerosis
- renal angiomyolipoma
- elevated serum vascular endothelial growth factor D (VEGF-D) of > 800 pg/mL
- thoracic or abdominal chylous effusion
- lymphatic malformations
- demonstration of LAM cells or LAM cell clusters on cytological examination of effusions or lymph nodes
- histopathological confirmation of LAM by lung biopsy or biopsy of retroperitoneal or pelvic masses
Pathology
The disease is characterized by the persistence of dilated lymphatics and interstitial proliferation of abnormal smooth muscle that in turn can obstruct venules, lymphatics, and small airways.
In the chest, there are two phases of proliferation in lymphangioleiomyomatosis. The early phase is characterized by proliferation of immature muscle cells that cover alveolar walls, bronchioles, pleura and vessels, including lymphatic routes. In the late phase, there is a development of cystic spaces and wider proliferation of muscle cells throughout the lung.
Radiographic features
Lymphangioleiomyomatosis is a multi-system disorder and can affect many organs.
Chest
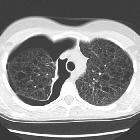
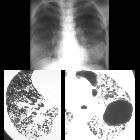
Multiple thin-walled cysts throughout the lungs, usually with a uniform distribution. These are present in nearly all cases. If the cysts are small, they may be seen as diffuse coarse interstitial markings on plain films.
- general/radiograph
- chylothorax: chylous pleural effusion
- evidence of hyperinflation
- diffuse bilateral reticulonodular densities
- recurrent pneumothoraces in complicated cases
- HRCT
- thin-walled cysts of variable sizes surrounded by normal lung parenchyma can be seen throughout the lung
- interlobular septal thickening
- may show a dilated thoracic duct
- hemorrhages may be seen as areas of increased attenuation
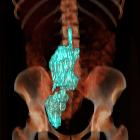
Abdomen and pelvis
- renal angiomyolipomas: most common abdominal finding
- splenic cysts
- chylous ascites
- lymphangioleiomyomas
- uterine fibroids
- abdominal lymphadenopathy
Neck
Bone
May show massive osteolysis with little or no periosteal reaction. Often multifocal disease.
Treatment and prognosis
Despite treatment with agents such as medroxyprogesterone and tamoxifen , LAM tends to be progressive with most of the disease severity due to pulmonary disease.
Complications
- recurrent pneumothorax can occur in up to 80% of cases
- thoracic chylous collections
- hemoptysis (occasional)
- pelvic lymphatic obstruction
Differential diagnosis
For pulmonary manifestations, the primary differential to be considered is:
- Langerhans cell histiocytosis (LCH)
- tends to happen in children and young adults with a history of heavy cigarette smoking
- mid to upper lobe distribution with preservation of the costophrenic angles
- cysts in LCH tend to be more irregular in contour
- has a much more favorable prognosis compared with LAM
- lymphocytic interstitial pneumonitis (LIP): smooth cysts with ground-glass attenuation and nodules, often associated with autoimmune disease
Siehe auch:
- Langerhanszell-Histiozytose der Lunge
- Lungenemphysem
- Tuberöse Sklerose
- Angiomyolipom der Niere
- multiple zystische Lungenherde
- Lymphangioleiomyom
und weiter:
- Bronchiektasen
- Angiomyolipom
- Kavernöse Lungenläsionen
- Lungenzysten
- Chylothorax
- Pneumocystis jiroveci Pneumonie
- Lymphangiom
- causes of pulmonary arterial hypertension
- retikuläres Muster
- PLCH
- Emphysemtypen
- Emphysem
- Chyloperitoneum
- pulmonary manifestations of cystic fibrosis
- pulmonary arterial hypertension classification - third world symposium on PAH
- Leiomyomatose
- Pneumothorax und Fliegen
- pulmonary lymphangioleiomyomatosis and renal angiomyolipoma
- tuberous sclerosis diagnostic criteria
- tuberous sclerosis complex with lymphangiomyomatosis
- Lymphangiomyom des Mesenteriums
- pelvic lymphangioleiomyomatosis
- thin-walled lung cysts
- retroperitoneale Lymphangioleiomyomatose


 Assoziationen und Differentialdiagnosen zu Lymphangioleiomyomatose:
Assoziationen und Differentialdiagnosen zu Lymphangioleiomyomatose:


