Creutzfeldt-Jakob-Krankheit





















Creutzfeldt-Jakob disease (CJD) is a spongiform encephalopathy that results in a rapidly progressive dementia and death usually within a year from onset. The vast majority are sporadic, but familial and acquired forms are occasionally encountered.
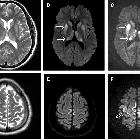
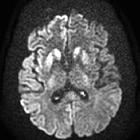
On imaging, it classically manifests as hyperintense signal on DWI (and usually FLAIR) in regions of the cerebral grey matter (cortex, followed by striatum, followed by thalamus).
Epidemiology
Four types of Creutzfeldt-Jakob disease have been described :
- sporadic (sCJD)
- accounts for 85-90% of cases
- further divided into numerous subtypes according to molecular markers (see pathology section below)
- variant (vCJD)
- bovine-to-human transmission of bovine spongiform encephalopathy (a.k.a. "mad cow disease"): considered zoonotic by some
- especially if in the UK between 1981-1996
- subsequent human-to-human transmission (e.g. transfusion) of vCJD
- familial (fCJD)
- 10% of cases
- these individuals carry a PRPc mutation
- iatrogenic (iCJD)
- following administration of cadaveric human pituitary hormones (pre-1985)
- various transplants
Clinical presentation
Sporadic Creutzfeldt-Jakob disease is characterized by rapidly progressive dementia and other features of neuropsychiatric decline resulting in death within a year of onset. Other common features include myoclonus, visual hallucinations, cerebellar dysfunction (such as ataxia and nystagmus), pyramidal or extrapyramidal signs (such as spasticity, rigidity, dystonia, or bradykinesia), and eventually akinetic mutism .
The Heidenhain variant presents as isolated visual symptoms such as impaired visual acuity, distortions of shapes and colors, and visual hallucinations.
The Brownell-Oppenheimer variant presents as isolated cerebellar ataxia.
Variant Creutzfeldt-Jakob disease presents mostly with psychiatric symptoms (such as depression) and sensory symptoms (dysesthesias or paresthesias).
Diagnostic markers
- characteristic results on electroencephalography (EEG)
- S100
- CSF 14-3-3 protein: positive result in patient suspected clinically of having sCJD
- CSF and/or olfactory mucosa real time quaking-induced conversion (RT-QuIC) seeding assays: detects minute amounts of the disease-specific pathologic prion protein
- found to be more sensitive than CSF 14-3-3 protein titers
A definitive diagnosis requires a brain biopsy, although in many institutions the difficulty involved in sterilizing equipment renders a biopsy undesirable.
Pathology
CJD is thought to be mediated via prions, a type of protein, which manifests in sheep as the disease scrapie, and in cows as bovine spongiform encephalopathy. Prions are considered infectious in the sense that they can alter the structure of neighboring proteins.
CJD leads to spongiform degeneration of the brain, which is thought to be caused by the conversion of normal prion protein to proteinaceous infectious particles that accumulate in and around neurons and lead to cell death.
Classification
A number of subtypes of sporadic CJD are recognized based on molecular markers, and these have distinct clinical and pathological features. The classification is based on :
- codon 129 in the prion protein gene
- methionine (M) or valine (V)
- size of the protease-resistant core of the abnormal prion protein
- PrPSc type 1 (21 kDa) or type 2 (19 kDa)
Each variant of sporadic CJD results from the combination of codon 129 genotype (M or V) and PrPSc type (1, 2 or 1 + 2) .
Additionally, MM2 subtype as has been noted to have distinctive histopathological features affecting the cerebral cortex and the thalamus and have therefore been separated into MM2 cortical (MM2C) and MM2 thalamic (MM2T).
Mixed types are also encountered. The result is that there is significant variation in the literature in regards to how many subtypes there are and if and how they should be grouped together .
Clinical patterns have been linked to various molecular subtypes (e.g. Heidenhain variant linked to MM1 and MM2C , and Brownell-Oppenheimer variant linked to VV2 ).
Radiographic features
Specific sites of imaging abnormality are typical of sporadic Creutzfeldt-Jakob disease :
- cerebral cortex (most common) :
- most common: insula, cingulate gyrus, superior frontal gyrus
- common: precuneus, cuneus, paracentral lobule, medial frontal gyrus, occipital gyri, angular/supramarginal gyrus, superior parietal lobule, inferior frontal gyrus
- less common: postcentral gyrus, precentral gyrus, medial and superior temporal gyri
- deep gray matter
Involvement is usually bilateral but may be asymmetric or symmetric . The extent of abnormality increases as the disease progresses.
The Heidenhain variant of sporadic Creutzfeldt-Jakob disease predominantly involves the parieto-occipital cortex.
Variant Creutzfeldt-Jakob disease characteristically shows the hockey stick sign and/or pulvinar sign of thalamic involvement. However, thalamic involvement is not pathognomonic of variant disease and is more commonly seen with sporadic Creutzfeldt-Jakob disease .
MRI
Cerebral gray matter findings are detected on the following sequences :
- DWI/ADC: restricted diffusion (most sensitive)
- T2-FLAIR: hyperintensity
Signal abnormalities may be subtle initially but become more pronounced as disease progresses. Review of sequential studies also typically demonstrates rapidly progressive cerebral atrophy.
An unusual finding associated with sporadic Creutzfeldt-Jakob disease is intrinsic T1 hyperintensity in the globus pallidus .
Cerebellar involvement, including in the Brownell-Oppenheimer variant, is primarily detected as atrophy; DWI is insensitive here .
Nuclear medicine
Fluorine-18-FDG PET shows hypometabolism in the affected regions.
Treatment and prognosis
There is currently no curative treatment and the disease is invariably fatal with a mean survival of only seven months.
Creutzfeldt-Jakob disease is a notifiable disease in most countries, including European Union countries, Australia, United Kingdom, USA, and Canada.
History and etymology
It was named after Hans Gerhard Creutzfeldt (1885-1964), a German neurologist who first described the condition in 1920, and Alfons Maria Jakob (1884-1931), a German neurologist who also described the condition in a separate study in 1921 .
Differential diagnosis
MRI imaging features overlap with many other conditions but the presentation will be different :
- autoimmune encephalitis
- particularly anti-D2 dopamine antibodies
- T2 signal change is dominant, with only minor if any diffusion change
- hypoxic/anoxic brain injury
- osmotic demyelination
- encephalitis
- hepatic encephalopathy
- hypoglycemic encephalopathy
- mitochondrial disease


 Assoziationen und Differentialdiagnosen zu Creutzfeldt-Jakob-Krankheit:
Assoziationen und Differentialdiagnosen zu Creutzfeldt-Jakob-Krankheit: