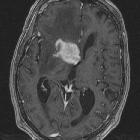
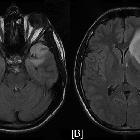
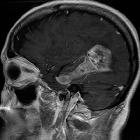
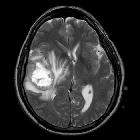
Glioblastoma multiforme

















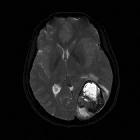







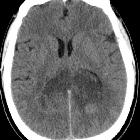

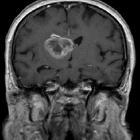










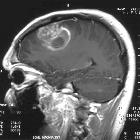


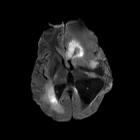





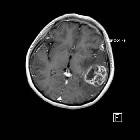

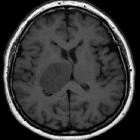
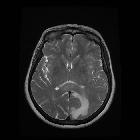















Glioblastoma (GBM) is the most common adult primary intracranial neoplasm (see brain tumors), accounting for 15% of all intracranial neoplasms and approximately 50% of all astrocytomas. GBMs are high-grade astrocytomas; they are therefore generally aggressive, largely resistant to therapy, and have a corresponding poor prognosis.
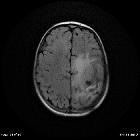
They have a predilection for spreading along the condensed white matter tracts such as corticospinal tracts and corpus callosum to involve the contralateral hemisphere.
Terminology
Glioblastoma was previously known as glioblastoma multiforme; the multiforme refers to the tumor heterogeneity. The WHO classification has dropped the 'multiforme' and thus it is best to refer to these tumors merely as glioblastomas. Somewhat confusingly the abbreviation GBM is still considered appropriate .
Primary vs secondary
Glioblastomas have traditionally been divided into primary and secondary; the former arising de novo (90%) whereas the latter developed from a pre-existing lower grade tumor (10%).
These correlate closely to IDH mutation status:
- IDH mutant: generally secondary glioblastoma, almost always MGMT methylated
- IDH wild-type: generally primary glioblastoma
If IDH status is unavailable or indeterminate then currently the diagnosis of glioblastoma NOS (not otherwise specified) should be made .
Primary
Primary glioblastomas are those that arise de novo, without a pre-existing lower grade diffuse astrocytoma. They account for 90% of all glioblastomas and are more aggressive than secondary glioblastomas and they tend to occur in older individuals.
Primary glioblastomas are almost invariably IDH wild-type. They tend to have amplification of EGFR and overexpression of MDM2, PTEN mutation and/or loss of heterozygosity of chromosome 10p .
Secondary
Secondary glioblastomas, in contrast, are those which arise from a pre-existing lower grade diffuse astrocytoma. They are relatively uncommon, only accounting for approximately 10% of all glioblastomas. These tumors tend to be less aggressive than primary glioblastomas and they tend to occur in younger patients . Interestingly, and of uncertain significance, they have a predilection for the frontal lobes .
Characteristically, and unlike primary tumors, secondary glioblastomas tend to be IDH mutant (positive), a mutation shared by over 80% of grade II and III astrocytomas . Secondary glioblastomas also demonstrate p53 mutations, amplification of PDGF-A, loss of heterozygosity of chromosomes 10q and 17p, loss of 19q and increased telomerase activity and hTERT expression .
Variants
In the current (2016) WHO classification of CNS tumors, three glioblastoma histological variants are recognized (which are discussed separately) as well as a number of histological patterns which are discussed below .
The three recognized variants are:
The remainder of this article concerns itself with primary (IDH wild-type) glioblastoma.
Epidemiology
A glioblastoma may occur at any age, however, they usually occur after the age of 40 years with a peak incidence between 65 and 75 years of age. There is a slight male preponderance with a 3:2 M:F ratio . Caucasians are affected more frequently than other ethnicities: Europe and North America 3-4 per 100,000 whereas Asia 0.59 per 100,000 .
The vast majority of glioblastomas are sporadic. Rarely they are related to prior radiation exposure (radiation-induced GBM). They can also occur as part of rare inherited tumor syndromes, such as p53 mutation related syndromes such as neurofibromatosis type1 (NF1) and Li-Fraumeni syndrome. Other syndromes in which GBMs are encountered include Turcot syndrome, Ollier disease and Maffucci syndrome.
Clinical presentation
Typically patients present in one of three ways:
- focal neurological deficit
- symptoms of increased intracranial pressure
- seizures
Rarely (<2%) intratumoral hemorrhage occurs and patients may present acutely with stroke-like symptoms and signs.
Pathology
Although glioblastomas can arise anywhere within the brain, they have a predilection for the subcortical white matter and deep grey matter of the cerebral hemispheres, particularly the temporal lobe .
Macroscopic appearance
Glioblastomas are typically poorly-marginated, diffusely infiltrating necrotic masses localized to the cerebral hemispheres. The supratentorial white matter is the most common location.
These tumors may be firm or gelatinous. Considerable regional variation in appearance is characteristic. Some areas are firm and white, some are soft and yellow (secondary to necrosis), and still others are cystic with local hemorrhage. GBMs have significant variability in size from only a few centimeters to lesions that replace a hemisphere. Infiltration beyond the visible tumor margin is always present.
These tumors are multifocal in 20% of patients but are rarely truly multicentric.
Microscopic appearance
Pleomorphic astrocytes with marked atypia and numerous mitoses are seen. Necrosis and microvascular proliferation are hallmarks of glioblastomas (see WHO grading of astrocytomas).
Microvascular proliferation results in an abundance of new vessels with a poorly formed blood-brain barrier (BBB) permitting the leakage of iodinated CT contrast and gadolinium into the adjacent extracellular interstitium resulting in the observed enhancement on CT and MRI respectively .
Edema and enhancement are however also seen in lower grade tumors that lack endovascular proliferation (anaplastic astrocytoma and other diffuse astrocytomas, for example, gemistocytic astrocytomas) and this is thought to be due to disruption of the normal blood-brain barrier by tumor produced factors. Vascular endothelial growth factor (VEGF) for example has been shown to both disrupt tight junctions between endothelial cells and increase the formation of fenestrations .
Cellular variants
Glioblastomas are capable of demonstrating varied patterns, sometimes within the one tumor. In addition to the three recognized variants (giant cell glioblastoma, gliosarcoma, and epithelioid glioblastoma) additional histological features are sometimes encountered which impact imaging appearance and biological behavior. Most of these are seen predominantly in primary IDH wild-type glioblastomas. These include :
- gemistocytes
- more commonly seen in secondary IDH mutant glioblastoma arising from a pre-existing gemistocytic astrocytoma
- granular cells
- histologically mimic macrophages and thus can lead to a misdiagnosis of macrophage-rich demyelination
- lipidized cells
- metaplasia
- most commonly squamous epithelium
- if dominant feature then a diagnosis of gliosarcoma should be considered
- multinucleated giant cells
- a common feature of glioblastoma
- if they are the dominant feature then a diagnosis of giant cell glioblastoma should be considered
- oligodendroglioma component
- must be either IDH wild-type or IDH mutant but 1p19q intact
- if IDH mutant and 1p19q co-deleted then regardless of other histological features it represents an anaplastic oligodendroglioma (WHO grade III)
- primitive neuronal cells
- previously known as glioblastoma with PNET-like component
- more frequently has CSF spread
- MYC or MYCN amplification common
- IDH mutant in 15-20% of cases
- small cell glioblastoma
- histologically appears similar to oligodendroglioma cell, but are IDH wild-type and commonly usually demonstrate EGFR amplification
- like oligodendrogliomas, they have a predilection for extensive cortical involvement
Immunophenotype
- GFAP: positive but of variable intensity
- S100: positive
- nestin: positive
- p53 protein: positive if TP53 mutated
- EGFR: positive in 40-98% of cases
- IDH-1 R132H: negative (by definition, otherwise not an IDH wild-type GBM, but rather a secondary IDH mutant tumor)
- H3 K27M mutation: negative (if positive then diffuse midline glioma H3 K27M-mutant)
Genetics
As discussed above, the vast majority of glioblastomas are primary and are IDH wild-type. IDH mutations are more common, and perhaps synonymous of, secondary glioblastomas (those arising from a pre-existing lower grade diffuse astrocytoma) .
TERT promoter mutations are frequently encountered and have a negative impact on prognosis, not as pronounced, however, as on lower grade diffuse astrocytomas .
Radiographic features
Glioblastomas are typically large tumors at diagnosis. They often have thick, irregular-enhancing margins and a central necrotic core, which may also have a hemorrhagic component. They are surrounded by vasogenic-type edema, which in fact usually contains infiltration by neoplastic cells.
Multifocal disease, which is found in ~20% of cases, is that where multiple areas of enhancement are connected to each other by abnormal white matter signal, which represents microscopic spread to tumor cells. Multicentric disease, on the other hand, is where no such connection can be seen.
CT
- irregular thick margins: iso- to slightly hyperattenuating (high cellularity)
- irregular hypodense center representing necrosis
- marked mass effect
- surrounding vasogenic edema
- hemorrhage is occasionally seen
- calcification is uncommon
- intense irregular, heterogeneous enhancement of the margins is almost always present
MRI
- T1
- hypo to isointense mass within white matter
- central heterogeneous signal (necrosis, intratumoral hemorrhage)
- T1 C+ (Gd)
- enhancement is variable but is almost always present
- typically peripheral and irregular with nodular components
- usually surrounds necrosis
- T2/FLAIR
- hyperintense
- surrounded by vasogenic edema
- flow voids are occasionally seen
- GE/SWI
- susceptibility artefact on T2* from blood products (or occasionally calcification)
- low-intensity rim from blood product
- incomplete and irregular in 85% when present
- mostly located inside the peripheral enhancing component
- absent dual rim sign
- DWI/ADC
- solid component
- elevated signal on DWI is common in solid/enhancing component
- diffusion restriction is typically intermediate similar to normal white matter, but significantly elevated compared to surrounding vasogenic edema (which has facilitated diffusion)
- ADC values correlate with grade
- WHO IV (GBM) = 745 ± 135 x 10 mm/s
- WHO III (anaplastic) = 1067 ± 276 x 10 mm/s
- WHO II (low grade) = 1273 ± 293 x 10 mm/s
- ADC threshold value of 1185 x 10 mm/s sensitivity (97.6%) and specificity (53.1%) in the discrimination of high-grade (WHO grade III & IV) and low-grade (WHO grade II) gliomas
- non-enhancing necrotic/cystic component
- the vast majority (>90%) have facilitated diffusion (ADC values >1000 x 10 mm/s)
- care must be taken in interpreting cavities with blood product
- solid component
- MR perfusion: rCBV elevated compared to lower grade tumors and normal brain
- MR spectroscopy
- typical spectroscopic characteristics include
- choline: increased
- lactate: increased
- lipids: increased
- NAA: decreased
- myoinositol: decreased
- typical spectroscopic characteristics include
PET
PET demonstrates the accumulation of FDG (representing increased glucose metabolism) which typically is greater than or similar to metabolism in grey matter.
Radiology report
When reporting a new diagnosis of a mass that is likely a glioblastoma, it is useful to include:
- morphology
- size in three dimensions
- degree of central necrosis
- non-enhancing tumor involving cortex, deep grey or white matter: look at ADC for lower values
- presence of necrosis
- relationship to/involvement of
- eloquent areas
- major white matter tract
- large vessels
- extension
- across midline
- into brainstem
- subependymal spread
- CSF dissemination
Treatment and prognosis
Biopsy and tumor debulking with postoperative adjuvant radiotherapy and chemotherapy (temozolomide) are the most commonly carried out treatment. Newer therapies include antiangiogenesis (e.g. bevacizumab) and immunotherapy.
In individuals 70 years of age or younger standard Stupp protocol is usual. In older individuals, radiotherapy is usually administered as a shorter course, but even in this setting adding temozolomide significantly increases survival, especially in MGMT methylated (inactive) tumors .
Despite this, it carries a poor prognosis with a median survival of fewer than 2 years .
Negative prognostic factors include:
- the degree of necrosis
- the degree of enhancement
- deep location (e.g. thalamus)
- MGMT not-methylated
- increased age
- lower pre-diagnosis functional status (e.g. ECOG performance status)
Followup
Glioblastomas are generally followed up fairly closely with MRI. Although timing and frequency will vary between institutions and treating surgeons/oncologists, generally a scan is obtained within 24-48 hours of surgery to assess residual disease (before postoperative enhancement develops) and thereafter every 8 to 12 weeks. In individuals who have no residual macroscopic disease and remain stable for a protracted time, the frequency of follow-up imaging can be decreased.
The primary aims of follow up are:
- identify tumor progression and complications thereof
- distinguish tumor progression from pseudoprogression
- distinguish pseudoresponse from tumor progression
Response assessment criteria
Glioblastomas have been the subject of close trial scrutiny with many new chemotherapeutic agents showing promise. As such a number of criteria have been created over the years to assess response to treatment. Currently, the RANO criteria are most widely used. Other historical systems are worth knowing to allow interpretation of older data. These systems for response criteria for first-line treatment of glioblastomas include :
- RANO criteria (most commonly used today)
- Macdonald criteria
- AVAglio criteria
- RTOG 0825 criteria
History and etymology
The original term glioblastoma multiforme was coined in 1926 by Percival Bailey and Harvey Cushing; the suffix multiform was meant to describe the various appearances of hemorrhage, necrosis, and cysts.
Differential diagnosis
General imaging differential considerations include:
- cerebral metastasis
- may look identical
- both may appear multifocal
- metastases usually are centered on grey-white matter junction and spare the overlying cortex
- rCBV in the 'edema' will be reduced
- primary CNS lymphoma
- should be considered especially in patients with AIDS, as in this setting central necrosis is more common
- otherwise usually homogeneously enhancing
- cerebral abscess
- central restricted diffusion is helpful, however, if GBM is hemorrhagic then assessment may be difficult
- presence of smooth and complete SWI low-intensity rim
- presence of dual rim sign
- anaplastic astrocytoma
- should not have central necrosis
- consider histology sampling bias
- tumefactive demyelination
- can appear similar
- often has an open ring pattern of enhancement
- usually younger patients
- subacute cerebral infarction
- history is essential in suggesting the diagnosis
- should not have elevated choline
- should not have elevated rCBV
- cerebral toxoplasmosis


 Assoziationen und Differentialdiagnosen zu Glioblastoma multiforme:
Assoziationen und Differentialdiagnosen zu Glioblastoma multiforme: