Sichelzellenanämie


















Sickle cell disease (SCD) (historically known as drepanocytosis) is a hereditary (autosomal recessive) condition resulting in the formation of abnormal hemoglobin (a hemoglobinopathy), which manifests as multisystem ischemia and infarction, as well as hemolytic anemia.
Hemoglobin SC (HbSC) disease, although a sickle cell disease subtype, with similarities to the classic condition, should ideally be considered as a distinct pathological entity .
Terminology
The term sickle cell disease is preferred to sickle cell anemia for the name of the condition, not least because the former term reflects the fact that the condition has multisystem effects, rather than just a severe form of anemia.
Epidemiology
There is no recognized gender predilection. The highest incidence occurs in individuals of African descent, followed by eastern Mediterranean and Middle Eastern populations. Malaria is the strongest known selective pressure on the human genome. The sickle cell mutation is prevalent in part as it confers a human genetic resistance to malaria as the abnormal hemoglobin has higher turnover and increased phagocytosis while sickled red cells have reduced cell-cell cytoadherence preventing the parasite from multiplying during the erythrocytic phase of its life cycle. It is estimated that approximately 8% of the African population is homozygous for sickle cell (where malaria is most prevalent).
Clinical presentation
The earliest manifestation is usually in early childhood, as babies are protected by elevated levels of fetal hemoglobin (HbF) in the first 6 months . The first presentation is commonly a painful vaso-occlusive crisis: sudden onset of bone or visceral pain due to microvascular occlusion and ischemia, often in the setting of sepsis or dehydration. Sickle cell disease is known to have a wide spectrum of clinical presentations from completely asymptomatic to a severe overwhelming crisis.
Clinical findings are wide and include :
- bone pain
- bone infarction
- subperiosteal hemorrhage
- osteomyelitis
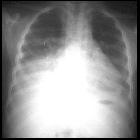
- pulmonary
- acute chest syndrome
- recurrent pneumonia (impaired immunity due to functional asplenia)
- chronic lung disease
- abdominal
- abdominal pain from vaso-occlusive crises
- sequestration syndrome (rapid pooling of blood in the spleen leading to intravascular volume depletion)
- hemolytic anemia and extramedullary hematopoiesis
- impaired immunity from autosplenectomy
- multiple renal manifestations, with end result of renal failure
- cerebral
- stroke
- cognitive impairment
- ocular and orbital complications
- priapism
- leg ulcers
Pathology
The disease results from a mutation in a gene coding for the beta chain of the hemoglobin molecule termed HbS. Specifically, there is a substitution of glutamine for valine at the 6th position in the beta-globin chain.
The term "sickle cell disease" applies to all patients who have two abnormal beta chains. The resultant hemoglobin molecules tend to clump together into long polymers, making the red blood cell (RBC) elongated (sickle-shaped), rigid and unable to deform appropriately when passing through small vessels, resulting in vascular occlusion. The abnormal RBCs are also removed from the bloodstream at an increased rate, leading to a hemolytic anemia .
Individuals with one HbS beta chain and one normal beta chain are said to have the "sickle cell trait". They are usually asymptomatic, although there is an association with an increased risk of renal medullary carcinoma . Perhaps of some consolation to individuals with the sickle cell trait is the increased resistance to malaria.
Individuals with one HbS beta chain and one hemoglobin C (HbC) beta chain, have a subtype of sickle cell disease known as hemoglobin SC (HbSC) disease .
Radiographic features
The radiographic manifestations of sickle cell disease are protean and are best discussed individually. Below is a summary of the main findings with links to individual articles.
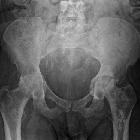
Skeletal
Musculoskeletal manifestations of sickle cell disease are discussed separately. Three separate mechanisms can result in skeletal changes :
These, in turn, can predispose individuals to other complications, such as growth disturbance and pathological fractures.
Pulmonary
Pulmonary involvement is a leading cause of mortality among sickle cell disease patients and can be acute or chronic:
Abdominal
Abdominal manifestations of sickle cell disease are discussed separately. Splenic infarction and subsequent functional asplenia tend to occur early in the disease. The hepatobiliary and renal systems are also commonly involved.
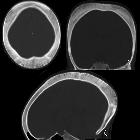
Cerebral
Cerebral manifestations of sickle cell disease are discussed separately. Stroke and cerebral atrophy are common neurologic sequelae of sickle cell disease.
Extramedullary hematopoiesis
Extramedullary hematopoiesis is discussed separately. Less common in sickle cell disease than in other hemolytic anemias. The most common site is liver, followed by spleen, thorax, and adrenals .
Treatment and prognosis
Management of vaso-occlusive crises includes oxygen, hydration, and analgesia. Hydroxyurea decreases the severity of vaso-occlusive crises . Anemia is usually well-tolerated, however, blood transfusions may be indicated in some cases. Bone marrow transplantation may provide a cure.
Sickle cell disease is associated with reduced life expectancy, whereas individuals with sickle cell trait have a normal life expectancy.
See also
Siehe auch:
- Osteomyelitis
- extramedulläre Hämatopoese
- Knocheninfarkt
- Milzinfarkt
- Thalassämie
- Autosplenektomie
- medulläres Nierenkarzinom
- muskuloskelettale Manifestationen bei Sichelzellanämie
- Acute chest syndrome bei Sichelzellanämie
- Sichelzellenanämie (abdominelle Manifestationen)
- Sichelzellenanämie (zerebrale Manifestationen)
- Sichelzellenanämie (chronische Lungenerkrankung)
- chronic lung disease
- dense bone mnemonic
und weiter:
- Aseptische Knochennekrose
- diffuse bony sclerosis (mnemonic)
- avascular necrosis causes (mnemonic)
- Nierenzellkarzinom
- Splenomegalie
- coarsened hepatic echotexture
- Osteomyelofibrose
- medulläre Nephrokalzinose
- Gamna-Gandy Körperchen
- codfish vertebra
- renal papillary necrosis (mnemonic)
- Kardiomegalie
- Moyamoya-Erkrankung
- FCE
- Auftreibung Metaphysen
- dichte metaphysäre Bänder
- Netzhautablösung
- Bürstenschädel
- alternating radiolucent and radiodense metaphyseal lines
- Budd-Chiari-Syndrom
- inanimate object inspired signs
- Knochenmark
- sausage digit
- stroke in children and young adults
- Verkürzung Metakarpale vier oder fünf
- premature closure of a growth plate
- Knochen-in-Knochen-Aspekt
- diffuse skelettale Sklerosierung
- H-förmige Wirbel
- Knochenmarksexpansion
- echogenic renal pyramids
- Moyamoya-Muster
- Hereditäre Sphärozytose
- papillary necrosis (mnemonic)
- transverse metaphyseal lines (mnemonic)
- Paroxysmale nächtliche Hämoglobinurie
- low signal intensity renal parenchyma
- akuter Verschluss Arteria renalis
- acute bone infarct due to sickle cell anemia
- H-shaped vertebrae in patient with sickle cell anemia
- intraabdominelle Verkalkungen
- Sichelzellenanämie Wirbelsäule
- step off vertebrae
- chronische Anämie
- signalarme Nieren
- acute splenic sequestration
- avascular necrosis in sickle cell disease
- sickle cell crisis
- Sichelzellenanämie Milz
- cerebral infarction in sickle cell disease


 Assoziationen und Differentialdiagnosen zu Sichelzellenanämie:
Assoziationen und Differentialdiagnosen zu Sichelzellenanämie:






