zystische Fibrose



























Cystic fibrosis (CF), also called mucoviscidosis, is an autosomal recessive genetic disease that affects the exocrine function of the lungs, liver, pancreas, small bowel, sweat glands, and the male genital system . resulting in progressive disability and multisystem failure. This article is a general discussion of the disease. Each organ system will be discussed separately.
- pulmonary manifestations
- bronchiectasis
- pneumothorax
- recurrent bacterial infection
- pulmonary arterial hypertension
- abdominal manifestations
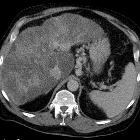
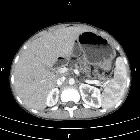
- pancreas (most common abdominal organ involved )
- exocrine and endocrine insufficiency
- fatty replacement of pancreas
- pancreatitis (acute and chronic)
- pancreatic cysts
- liver
- hepatic steatosis
- focal biliary and multilobular cirrhosis
- portal hypertension
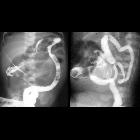
- biliary system
- gastrointestinal tract
- distal intestinal obstruction syndrome (DIOS)
- meconium ileus: 10-20%
- rectal prolapse
- esophageal dysfunction / gastro-esophageal reflux
- distension of appendix but reduced risk of appendicitis
- pancreas (most common abdominal organ involved )
- head and neck manifestations
- musculoskeletal manifestations
- urogenital tract manifestations
- bilateral vesicle seminal agenesis
- testicular microlithiasis
- hypoplasia or agenesis of the vas deferens
- hypoplasia or agensis of the tail and body of the epididymis
Epidemiology
Cystic fibrosis is the most common genetic disease affecting European population with an incidence of approximately 1:2000-3500 live births .
Clinical presentation
The diagnosis may be suspected antenatally due to the presence of echogenic bowel on antenatal ultrasound, or due to genetic testing of the parents.
In many countries, the presence of cystic fibrosis is tested for immediately after birth with a sweat test (positive sweat chloride test Cl >60 mEq/L) . Alternatively, genetic testing is also available.
The diagnosis usually becomes evident in infancy, with presentations including:
- meconium ileus
- rectal prolapse
- recurrent pulmonary infection
Pathology
Genetics
Cystic fibrosis is due to a homozygous defect of the CFTR gene located on chromosome 7q31.2. This gene encodes for a transmembrane protein known as cystic fibrosis transmembrane regulator (CFTR) which is responsible for regulating chloride passage across cell membranes. There are at least 6 classes of mutations, the commonest being ∂F508 (66-70%) .
In skin, unlike elsewhere, the CFTR protein is responsible for the influx of chloride and increases the sodium channel activity, thus controlling the influx of sodium. The net effect of a normally functioning CFTR is to resorb sodium and chloride. In CF patients, this is lost and therefore the characteristic increase in salt content of sweat (thus the sweat test).
In tissues other than skin, the CFTR protein is responsible for efflux of chloride and inhibition of the sodium channel's activity which controls the influx of sodium. Therefore, under normal circumstances, salt and chloride remain in the lumen and keep water there osmotically. In CF patients, too little chloride is pumped out, too much sodium is resorbed with osmotic resorption of water from the lumen. The result is iso-osmotic, but low volume, secretions, which tend to dry out, or be thick as they still contain all the other constituents.
Treatment and prognosis
Early institution of multidisciplinary treatment is essential and responsible for the dramatic increase in life expectancy, now reaching 40 or more years.
Treatment options include :
- prolonged courses of antibiotics
- oral and inhaled corticosteroids
- pancreatic enzyme supplementation: required in 85% cases
- vitamin supplementation
- insulin
- physiotherapy
- lung transplantation
In the 2010s, a new group of drugs specifically targetting the molecular defects in cystic fibrosis known as CFTR modulators were introduced. These specifically act on the defective cystic fibrosis transmembrane regulator (CFTR) .
- ivacaftor: most effective in those with G551D defect, only 2.3% patients
- lumacaftor: designed for the common ∂F508 mutation, but little functional improvements in trials
- lumacaftor and ivacaftor: combination therapy: approved for ∂F508 mutations
Both transplanted and non-transplanted CF patients are at increased risk of some malignancies :
- gastrointestinal malignancy, in particular esophageal, gastric, small bowel, and colorectal cancer
- gallbladder and extrahepatic biliary tree
- lymphoma/leukemia
- testicular cancer
Siehe auch:
- Pneumothorax
- Bronchiektasen
- Leberzirrhose
- Steatosis hepatis
- Mekoniumileus
- pulmonale Hypertonie
- distal intestinal obstruction syndrome (DIOS)
- abdominelle Manifestationen bei Mukoviszidose
- pulmonale Manifestationen der Mukoviszidose
- echogenic bowel
- Cystic Fibrosis Transmembrane Regulator
und weiter:
- bronchial wall thickening
- mosaikartige Verdichtungen der Lunge
- Lungenfibrose
- Kartagener-Syndrom
- Pneumatosis coli
- Mukoidimpaktion
- zystische Pankreasläsionen
- tree in bud-Muster
- zentraler Venenkatheter
- Chronisch obstruktive Lungenerkrankung
- Ileumatresie
- dichte metaphysäre Bänder
- primäre ciliäre Dyskinesie
- causes of pulmonary arterial hypertension
- Pankreasverkalkungen
- Pneumoperitoneum beim Neugeborenen
- Gefäßverkalkungen
- Mekoniumperitonitis
- Differenzialdiagnosen bei Tracheomalazie
- brasfield scoring system
- bronchial arterial aneurysm
- Pankreaszyste
- upper lobe bronchiectasis
- Shwachman-Diamond syndrome
- Pneumatosis cystoides des Kolons
- central bronchiectasis
- DIOS
- pulmonary manifestations of cystic fibrosis
- Bronchozele
- upper lobe pulmonary fibrosis (mnemonic)
- Lubani-al-Saleh-Teebi-Syndrom
- non visualisation of the fetal gallbladder
- mucoid impaction - cystic fibrosis
- small bowel involvement with cystic fibrosis
- bronchiectasis (mnemonic)
- cystic fibrosis transmembrane regulator (CFTR)
- finger clubbing in a patient with cystic fibrosis
- pancreatic calcification with cystic fibrosis
- distal intestinal obstruction syndrome (DIOS) in cystic fibrosis
- Pankreasbeteilgung bei Mukoviszidose
- Mukoviszidose / Lungen-Diagnostik mit MRT
- Zystische Fibrose Einteilung


 Assoziationen und Differentialdiagnosen zu zystische Fibrose:
Assoziationen und Differentialdiagnosen zu zystische Fibrose: