spinal paraganglioma








Spinal paragangliomas are tumors of neuroendocrine origin that rarely involve the central nervous system, usually the filum terminale and cauda equina. They are indolent and considered WHO grade I lesions .
Paragangliomas overall are most commonly located within the adrenal gland (pheochromocytomas) or in the head and neck. For a general discussion of paragangliomas, please refer to parent article: paraganglioma
Epidemiology
The age range at presentation for spinal paragangliomas varies childhood to older age (9-75 years of age) with most cases being diagnosed in middle age (30-60 years) . Males are somewhat more commonly affected than females .
Spinal paragangliomas are sometimes seen in the context of Carney triad or Carney Stratakis syndrome.
Clinical presentation
The clinical presentation is either with local mass effects or neuroendocrine symptoms.
The most common presenting symptoms are lower back pain and sciatica from mass effect.
Cauda equina paragangliomas frequently actively secrete neuropeptides, particularly 5-hydroxytryptamine and somatostatin, although symptoms related to this chemical production are usually absent.
Additionally, these lesions may be a rare source of superficial siderosis, and thus present with sensory neural impairment, cranial nerve dysfunction and myelopathy .
Pathology
Paragangliomas of the spine usually arise in the region of the cauda equina, although rare reports of lesions elsewhere along the spine are available. They are indolent and considered WHO grade I tumors under the current (2016) WHO classification of CNS tumors .
Macroscopic appearance
Spinal paragangliomas are highly vascular masses, the majority (75%) of paragangliomas are encapsulated, usually attached to the filum terminale or less commonly a nerve root .
Microscopic appearance
All paragangliomas consist of two types of cells; type I and type II. The main components are lobules or nests of chief cells (type I); these structures are known as Zellballen. They are surrounded by a single layer of sustentacular cells (type II) .
Immunophenotype
Immunohistochemical examination confirms neuroendocrine differentiation of chief cells (type I) :
- chromogranin-A: positive
- synaptophysin: positive
Sustentacular cells (type II):
Genetics
Numerous mutations have been identified as predisposing to paragangliomas including: VHL, RET, NF1, SDHAF2, SDHB, SDHC, SDHD, TNEM127 and MAX .
Radiographic features
Plain film
Plain films are unlikely to demonstrate any abnormality, although vertebral scalloping or rarely calcification may be seen .
Angiography
Conventional angiography demonstrates an intense early blush that persists into the late arterial and early venous phases.
CT
Spinal paragangliomas appear as soft tissue masses inferior to the conus. If contrast is administered they enhance vividly (as do paragangliomas elsewhere).
Large lesions may rarely demonstrate osseous erosion or remodeling of the adjacent vertebrae (vertebral scalloping). Rarely these lesions calcify .
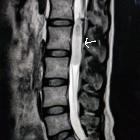
MRI
Similarly, on MRI these masses usually appear as well-circumscribed masses, inferior to the conus.
With some lesions, the characteristic “salt-and-pepper” appearance of neck and skull base paragangliomas may also be seen. Associated syringohydromyelia has been reported in some cases.
Reported signal characteristics include:
- T1: isointense
- T2
- hyperintense
- flow voids are typically seen along the surface of and within the tumor nodule
- hemorrhage is common, leading to a hemosiderin cap sign
- T1 C+ (Gd): intense enhancement is virtually always seen
Treatment and prognosis
Surgical resection is the treatment of choice, sometimes with preoperative embolization to reduce intra-operative blood loss. Resection alone is usually curative with a post-resection recurrence rate of less than 5% .
Rarely CSF dissemination is encountered .
Differential diagnosis
The differential diagnosis is essentially that of other intradural extramedullary tumors, particularly those located in the lumbar region.
- myxopapillary ependymoma
- neurogenic tumor
- spinal meningioma (rare in the lumbar region)
Siehe auch:
- spinale Schwannome
- spinal neurofibroma
- intraspinales Meningeom
- spinales Ependymom des Filum terminale
- intradurale extramedulläre Tumoren
- neurogenic tumour
und weiter:
- Schwannom
- Paragangliom
- WHO-Klassifikation der Tumoren des zentralen Nervensystems
- Cauda equina Lipom
- scalloping Wirbelkörper
- neoplasms of the spinal canal
- spinales Hämangioblastom
- spinal myxopapillary ependymoma
- spinales Ependymom
- Paragangliom des Nervus vagus
- Neuroforamen Zyste
- Paragangliome von Kopf und Hals
- solitärer fibröser Tumor des Rückenmarks
- Paragangliom an der Cauda equina


 Assoziationen und Differentialdiagnosen zu spinal paraganglioma:
Assoziationen und Differentialdiagnosen zu spinal paraganglioma: