glomus tumour



































 ähnliche Suchen
ähnliche Suchen siehe auch
siehe auchGlomus caroticum Tumor
Glomus jugulare Tumor
Phäochromozytom
spinal paraganglioma
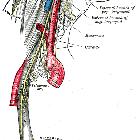
carotid bifurcation
Harnblase
glomangioma
carotid body
Nervus glossopharyngeus
Foramen jugulare
glomus body
glomus tympanicum tumour
pulssynchrones Ohrgeräusch
Fisch Klassifikation Glomus Tumor
Glomustumor am Finger
non chromaffin paraganglion cells
Klassifikation Glomus caroticum Tumor
Paragangliom des Nervus vagus
Vernet's syndrome
salt and pepper
Paragangliome von Kopf und Hals
Paragangliom des Herzens
nonchromaffin paraganglion cells
Zuckerkandl-Organ
Arnold nerve
Jacobson-Anastomose
Paragangliomas, sometimes called glomus tumors, are rare neuroendocrine tumors arising from paraganglia.
Terminology
Paraganglia are clusters of neuroendocrine cells dispersed throughout the body and closely related to the autonomic nervous system, with either parasympathetic or sympathetic function. The largest cluster of cells is found within the adrenal medulla, with smaller collections in the paravertebral space, and head and neck region. Tumor arising in the paraganglia are called paragangliomas. They are classified by both location and secretory function.
Clinical presentation
Sympathetic paragangliomas present with features of catecholamine-excess, such as headaches, palpitations, diaphoresis and hypertension. Whereas, parasympathetic paragangliomas present more commonly with mass-effect such as cranial nerve palsies, a neck mass or tinnitus.
Pathology
All paragangliomas consist of two types of cells; type I and type II. The main components are lobules or nests of chief cells (type I); these structures are known as Zellballen. They are surrounded by a single layer of sustentacular cells (type II) .
Malignancy is defined as evidence of metastases. Histologically there are no reliable markers of malignant potential. Most common sites for malignancy include lymph nodes, liver, lung, and bone.
Risk of malignancy:
- pheochromocytoma: 10%
- sympathetic paraganglioma: 20%
- parasympathetic panganglioma: 2-20%
It is important to note biopsy (incision or fine needle aspirate) is contraindicated in suspected paragangliomas until biochemical screening is negative for catecholamine excess, due to the risk of catecholamine crisis and severe hypertension.
Location
Parasympathetic paragangliomas
Parasympathetic paragangliomas arise within paraganglia of the head and neck (see - paragangliomas of the head and neck) in association with the branches of the glossopharyngeal and vagus nerve . They are generally non-secretory.
- carotid body paraganglioma (see carotid body tumor)
- jugulotympanic paraganglioma
- vagal paraganglioma
- laryngeal paraganglioma
Sympathetic paragangliomas
Sympathetic paragangliomas generally arise in paraganglia below the level of the neck. They tend to secrete catecholamines and can be intra- or extra-adrenal.
- extra-adrenal: arise outside the adrenal gland along the length of the sympathetic chain
- abdomen
- organ of Zuckerkandl
- bladder base
- thorax (see mediastinal paraganglioma)
- paravertebral (aortosympathetic paraganglia)
- great vessels of the chest (aortopulmonary paraganglia)
- cardiac (extremely rare; may be located along the epicardium, in the atrial cavity, the interatrial septum or the ventricles)
- abdomen
- intra-adrenal: arise within the adrenal medulla
Immunophenotype
Immunohistochemical examination confirms neuroendocrine differentiation of chief cells (type I):
- neuron-specific enolase (NSE)
- chromogranin-A
- synaptophysin
Sustentacular cells (type II):
Genetics
Paragangliomas are the most strongly hereditary group of tumors. The most common genetic cause of hereditary paragangliomas are mutations in the succinate dehydrogenase (SDH) subunit (SDHB, SDHD, SDHA or SDHAF2) .
They are also associated with four clinical syndromes:
- von Hippel-Lindau syndrome
- multiple endocrine neoplasia types 2A and 2B
- neurofibromatosis type 1
- Carney-Stratakis syndrome
von Hippel-Lindau syndrome and neurofibromatosis type 1 are more commonly associated with pheochromocytomas. SDH mutations are common in head and neck paragangliomas, except for SDHB, which is associated with sympathetic paragangliomas. SDHB also confers a higher risk of malignancy .
Radiographic features
Both anatomical and functional imaging of paragangliomas is required for diagnosis and staging. Anatomical imagining includes CT and MRI. Multiple functional imaging modalities exist: I-MIBG scintigraphy, F-FDA PET, F-DOPA PET, F-FDG PET and Ga-DOTATATE- PET.
CT and MRI are the initial imaging modalities for tumor localization. They have excellent sensitivity but lack specificity in unequivocally identifying a mass as a paraganglioma.
CT
- density greater than 10 HU on non-contrast imaging (differentiates from adenoma)
- avidly enhances with contrast with delayed washout (due to rich capillary network)
- can detect lesions 0.5 cm in diameter
MRI
- T1
- hypointense to liver and adrenal
- salt and pepper appearance due to enhancing parenchyma (salt) and signal flow voids of vessels (pepper)
- T2
- hyperintense 'light bulb' appearance
- salt and pepper enhancement also seen
- T1 C+ (Gd): heterogenous prolonged enhancement
Nuclear medicine
Targets for functional imaging:
- tumor-specific catecholamine production: I-MIBG, F-FDA and F-DOPA PET
- glucose: F-FDG
- somatostatin receptor (overexpressed in paragangliomas): [Ga]-DOTATATE-PET
Each modality has strengths and weaknesses in detecting lesions depending on their location, secretory function and underlying genetic mutation.
- I-MIBG
- strength: pheochromocytomas and extra-adrenal sympathetic paragangliomas
- weakness: head and neck, malignant disease, MEN2
- F-DOPA PET
- strength: head and neck paragangliomas, SDHD-mutations, non-metastatic disease
- weakness: SDHB mutations
- F-FDA
- strength: metastatic disease, non-metastatic pheochromocytomas
- weakness: limited clinical availability
- F-FDG PET
- strength: malignancy, SHDB-mutations, von Hippel-Lindau syndrome
- weakness: may lack specificity
- Ga-DOTATATE PET
- strength: overall viable imaging modality; proven superiority in sporadic disease, SHDB-mutations, head and neck lesions
- weakness: detection of liver and lung lesions
Treatment and prognosis
Treatment may include surgical resection or radiotherapy. Paragangliomas may metastasize to other locations.
Differential diagnosis
Differential diagnosis differs depending on the location and histology of the paraganglioma but can include :
- melanoma
- mesenchymal tumors
- adrenal cortical tumors

 Assoziationen und Differentialdiagnosen zu Glomustumor:
Assoziationen und Differentialdiagnosen zu Glomustumor: