Marfan syndrome





















Marfan syndrome is a multisystem connective tissue disease caused by a defect in the protein fibrillin 1, encoded by the FBN1 gene. Cardiovascular involvement with aortic root dilatation and dissection is the most feared complication of the disease.
Epidemiology
The estimated prevalence is around 2-6 per 100,000 . There is no recognized gender or racial predilection.
Clinical presentation
Patients with Marfan syndrome may have the following symptoms and signs:
- general
- tall stature
- long arm span (often exceeding the height of the patient)
- joint laxity resulting in recurrent dislocations
- spine/skull
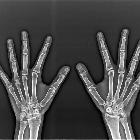
- hands
- arachnodactyly
- protrusion of thumb beyond fist when clenched (Steinberg sign)
- flexion deformity of the little finger
- pelvis / lower limbs
- chest wall deformities (present in up to ⅔ of cases )
- ocular
- ectopia lentis
- myopia
- cardiovascular
Patients may present with complications of the disease such as aortic dissection and pneumothorax.
Diagnostic criteria
The Ghent nosology was established in 1995 for the clinical diagnosis of the disease .
Pathology
Genetics
The condition results from a mutation in the fibrillin 1 (FBN1) gene located on chromosome 15q21.1 which is responsible for cross-linking collagen. In the majority of cases it is inherited in an autosomal dominant fashion, although in up to one-third of cases the mutation is de novo. The disease has high genetic penetrance but with variable phenotypic expression even amongst affected family members.
Studies show a regulatory relationship between extracellular microfibrils and TGFβ signaling, so an abnormality in either can cause a Marfanoid phenotype .
Microscopic appearance
Microscopically the arterial walls may show cystic medial necrosis .
Radiographic features
There are no specific radiographic features of Marfan syndrome but the following signs and complications of the disease may be seen in each system on a range of modalities:
Skeletal
- general
- osteopenia
- joint dislocation
- spine/skull
- atlantoaxial subluxation
- dural ectasia
- increased interpedicular distance
- kyphoscoliosis
- enlarged sacral foramina
- meningoceles
- presacral meningocele
- lateral sacral meningocele
- vertebral scalloping
- scaphocephaly
- pelvis / lower limbs
- progressive protrusio acetabuli
- slipped upper femoral epiphyses (SUFE)
- tibial subluxation
- patella alta
- pes planus
- hallux valgus
- clubfoot
- hands
- arachnodactyly
- protrusion of thumb beyond fist when clenched (Steinberg sign)
- flexion deformity of the little finger(s)
- chest wall deformities
Cardiovascular
- aortic root dilatation and myxomatous degeneration of the mitral valve resulting in mitral valve regurgitation are the most two common cardiac manifestations
- aortic aneurysm and aortic dissection are the most fearsome consequences - findings suggestive of higher risk of aortic dissection are:
- family history of aortic dissection
- dilatation at the aortic sinotubular junction
- aortic root diameter >55 mm
- increased aortic stiffness
- excessive aortic root dilatation (>1.7 mm/year)
- arterial dissection
- aortic valve regurgitation (AR)
- aortic coarctation
- pulmonary arterial dilatation
Pulmonary
- spontaneous pneumothorax
- lung cysts and bullae formation
Treatment and prognosis
Beta blockers are shown to reduce the rate of aortic root dilatation. In patients who can not take beta-blockers, calcium-channel blockers are used. More recently, investigators found that angiotensin receptor blockers which are also TGFβ antagonists significantly reduce cardiovascular and other somatic features . Cardiovascular complications are the most frequent cause of death .
Some of the more newer treatment options include.
History and etymology
First described in 1896 by Antoine Bernard-Jean Marfan, French pediatrician (1858-1942).
Differential diagnosis
- congenital contractural arachnodactyly: has significant phenotypic overlap with Marfan syndrome, but is now considered a discrete entity, due to distinct genetics
- Loeys-Dietz syndrome: similar features to Marfan syndrome
- homocystinuria: may resemble those with Marfan syndrome in some aspects;ectopia lentis, however, is downward as opposed to Marfan and intellectual disability is also a common feature
- multiple endocrine neoplasia type IIb may have a Marfan syndrome like body habitus
Siehe auch:
- Meningozele
- Duraektasie
- Loeys-Dietz-Syndrom
- erweiterter Spinalkanal
- Arachnodaktylie
- MEN IIb
- lateral meningocele
- Gorlin-Vickers-Syndrom
- Kyphoskoliose
- Kongenitale Kontrakturale Arachnodaktylie
- sakrale Meningozele
und weiter:
- Rippenusuren
- Ektasie Aorta ascendens
- Aneurysma
- Skoliose
- Dissektion Arteria vertebralis
- Bauchaortenaneurysma
- Protrusio acetabuli
- Liquorunterdrucksyndrom
- Kardiomegalie
- Klumpfuß
- scalloping Wirbelkörper
- Mikrognathie
- metacarpal index
- mandibuläre Retrognathie
- Nierenarterienaneurysma
- Aneurysma Aorta ascendens
- Aneurysma Koronararterien
- Karotisdissektion
- Dissektion
- Keratozystischer odontogener Tumor
- Hughes-Stovin-Syndrom
- Plattfuß
- dural ectasia of lumbo-sacral spine
- Aneurysma der Arteria pulmonalis
- stroke in children and young adults
- Differenzialdiagnosen bei Tracheomalazie
- femoropatellare Instabilität
- Zystische Medianekrose Erdheim-Gsell
- Erweiterung des interpedunculären Abstands
- idiopathisches Lungenemphysem mit riesigen Bullae
- dural ectasia associated with Marfan syndrome
- kongenitale Tracheomalazie
- gracile bones (mnemonic)
- differential of an enlarged pulmonary trunk on chest radiography
- keratocystic odentogenic tumour
- lumbosakrale Wirbelsäule bei Marfon-Syndrom
- erweiterte Pulmonalarterie
- Marfan - ICA dissection
- vermehrter Sagittaldurchmesser LWS
- anteriore sakrale Meningozele
- Aortenaneurysma bei Kindern und Jugendlichen
- lens dislocation related to Marfan syndrome
- Megalokornea
- Dilatation der Aorta thoracica


 Assoziationen und Differentialdiagnosen zu Marfan-Syndrom:
Assoziationen und Differentialdiagnosen zu Marfan-Syndrom:
