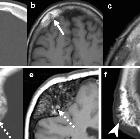
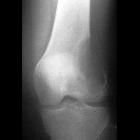
fibrous dysplasia























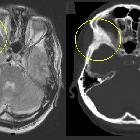




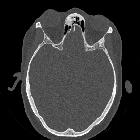
















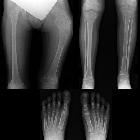

















 nicht verwechseln mit: osteofibröse Dysplasie
nicht verwechseln mit: osteofibröse DysplasieFibrous dysplasia (FD) is a non-neoplastic tumor-like congenital process, manifested as a localized defect in osteoblastic differentiation and maturation, with the replacement of normal bone with large fibrous stroma and islands of immature woven bone. Fibrous dysplasia has a varied radiographic appearance. If asymptomatic, it does not require treatment.
Terminology
Fibrous dysplasia can affect any bone and can be divided into four subtypes (although there is some overlap):
- monostotic: single bone
- polyostotic: multiple bones
- craniofacial fibrous dysplasia: skull and facial bones alone
- cherubism: mandible and maxilla alone (not true fibrous dysplasia)
The remainder of this article concerns itself with skeletal fibrous dysplasia. For a discussion of craniofacial fibrous dysplasia and cherubism, please refer to the respective articles.
Epidemiology
Fibrous dysplasia is found predominantly in children and young adults, with ~75% of patients presenting before the age of 30 years (highest incidence between 3 and 15 years). In polyostotic form, patients usually present by 10 years old. There is no recognized gender predilection .
Although fibrous dysplasia is usually sporadic, a number of associations are well recognized:
- McCune-Albright syndrome: in 2-3% of cases with the polyostotic form
- isolated endocrinopathy without the full McCune-Albright syndrome precocious puberty in girls
- hyperthyroidism
- hyperparathyroidism: renal stones, calcinosis
- acromegaly
- diabetes mellitus
- Cushing syndrome: osteoporosis, acne
- growth retardation
- Mazabraud syndrome: soft-tissue myxomas (rare); typically multiple intramuscular lesions in the vicinity of most severely affected bone
Clinical presentation
The condition is often an incidental finding and is usually painless. Alternatively, it may present due to bony expansion or remodeling. Morbidity may arise from compression and displacement of adjacent structures. This is particularly true in craniofacial fibrous dysplasia, where the content of the orbit or cranial nerves may be compressed.
Pathology
Fibrous dysplasia is due to developmental dysplasia and focal arrest in normal osteoblastic activity secondary to a non-hereditary mutation which results in the presence of all of the components of normal bone with a lack of normal differentiation into their mature structures.
Macroscopic appearance
Macroscopically, lesions are intramedullary and well circumscribed with abnormal whitish-grey color.
Microscopic appearance
Microscopically it manifests as large fibrous matrix with scattered curvilinear irregularly shaped trabeculae of immature, inadequately mineralized bone . There is no rimming by osteoblasts differentiating feature from cemento-ossifying fibroma. Cartilaginous islands are present in 10%, differentiating feature from chondrosarcoma.
Subtypes
Monostotic form (involving only one bone)
This is by far the most common and accounts for 70-80% of cases . It is usually asymptomatic until 2-3 decade but can be seen throughout adulthood . After puberty, the disease becomes inactive, and monostotic form does not progress to polyostotic form.
Polyostotic form
In the remaining 20-30% of cases, multiple bones are involved. As expected this presents earlier, typically in childhood (mean age of 8 years) with two-thirds symptomatic by the age of 10.
Location
Monostotic form
- ribs: 28%, most common
- proximal femur: 23%
- tibia
- craniofacial bones: 10-25%
- humerus
Polyostotic form
- often unilateral and monomelic: one limb
- femur: 91%
- tibia: 81%
- pelvis: 78%
- foot: 73%
- ribs
- skull and facial bones: 50%
- upper extremities
- lumbar spine: 14%
- clavicle: 10%
- cervical spine: 7%
Radiographic features
Plain radiograph
The appearance of fibrous dysplasia is usually smooth and homogenous with endosteal scalloping and cortical thinning . The borders are well defined and the cortex is usually intact but thinned due to the expansive nature of the lesion . Other features include:
- ground-glass matrix
- may be completely lucent (cystic) or sclerotic
- well circumscribed lesions
- no periosteal reaction
- rind sign
Pelvis and ribs
Ribs are the most common site of monostotic fibrous dysplasia. Fibrous dysplasia is the most common cause of a benign expansile lesion of a rib (see rib lesions)
- bubbly cystic lesions
- fusiform enlargement of ribs
- protrusio acetabuli
Extremities
- may lead to premature fusion of growth plates leading to short stature
- bowing deformities
- shepherd crook deformity of the femoral neck
- discrepant limb length
- Looser zones
CT
- ground-glass opacities: 56%
- homogeneously sclerotic: 23%
- cystic: 21%
- well-defined borders
- expansion of the bone, with intact overlying bone
- endosteal scalloping may be seen
MRI
MRI is not particularly useful in differentiating fibrous dysplasia from other entities as there is marked variability in the appearance of the bone lesions, and they can often resemble a tumor or more aggressive lesions.
- T1: heterogeneous signal, usually intermediate
- T2: heterogeneous signal, usually low, but may have regions of higher signal
- T1 C+ (Gd): heterogeneous contrast enhancement
Nuclear Medicine
Demonstrates increased tracer uptake on Tc bone scans (lesions remain metabolically active into adulthood).
Treatment and prognosis
Usually, no treatment is required as the bone lesions usually do not progress beyond puberty. If a mass effect is severe, then surgical decompression may be considered. The monostotic form does not transform or progress into the polyostotic form .
Complications
Not surprisingly, bone affected by fibrous dysplasia is weaker than normal and thus susceptible to pathological fractures.
Sarcomatous dedifferentiation (osteosarcoma [most common ], fibrosarcoma, malignant fibrous histiocytoma, or rarely chondrosarcoma) is occasionally seen (< 1%) and is more common in the polyostotic form. It should be noted that many reported cases may relate to previous treatment with radiation therapy .
Differential diagnosis
Due to the variability of the appearance of fibrous dysplasia the potential differential is very long but will be significantly influenced by the dominant pattern.
- Paget disease
- mosaic pattern bone histologically
- radiographically may be similar
- different demographics
- neurofibromatosis type I
- osseous lesions are rare
- vertebral column is the primary target
- ribbon ribs
- other features of the disease usually present
- osteofibrous dysplasia
- almost exclusively in tibia with anterior bowing
- lesion begins in cortex
- usually seen in children <10 years
- adamantinoma
- 80% seen in the tibia
- may appear indistinguishable
- non-ossifying fibroma
- simple bone cyst
- giant cell tumor
- enchondromatosis
- hemangioma
Siehe auch:
- Hämangiom Schädelkalotte
- Morbus Paget des Knochens
- nicht ossifizierendes Fibrom
- Neurofibromatose Typ 1
- einfache (juvenile) Knochenzyste
- Ollier-Syndrom
- kraniofaziale fibröse Dysplasie
- Cherubismus
- Riesenzelltumor
- Jaffé-Campanacci-Syndrom
- fibröse Dysplasie der Kalotte
- McCune-Albright Syndrom
- Fibröse Dysplasie der Rippen
- Adamantinom der langen Röhrenknochen
- Fibröse Dysplasie der langen Röhrenknochen
- maligne Entartung fibröse Dysplasie
- fibröse Dysplasie der Extremitätenknochen
- tibial fibrous dysplasia
- fibrous dysplasia in children
- Transformation der Knochenmatrix bei Fibröser Dysplasie
- fibröse Dysplasie des Beckens
und weiter:
- endosteal scalloping
- Hyperostosis frontalis interna
- Aneurysmatische Knochenzyste
- Ameloblastom
- Café-au-lait-Fleck
- dentigerous cyst
- Ossifizierendes Fibrom
- Adamantinom
- Hüftkopfnekrose
- idiopathische kindliche Hüftkopfnekrose
- Loosersche Umbauzonen
- Auftreibung Metaphysen
- Türflügelzeichen
- Tumoren der Thoraxwand
- Knochentumoren
- teleangiektatisches Osteosarkom
- conditions involving skin and bone
- liposklerosierender myxofibroider Tumor
- button sequestrum
- Hyperostosis Kalotte
- basilar invagination (mnemonic)
- arrested pneumatization of skull base
- polyostotische fibröse Dysplasie
- lytic bone lesion (mnemonic)
- zystische Angiomatose
- fibröser Kortikalisdefekt
- differential diagnosis for calcified masses in the mandible
- multiple benign lucent bone lesions (mnemonic)
- Fibröse Dysplasie der Schädelbasis
- focal sclerotic bony lesions (mnemonic)
- lytische Läsionen der Mandibula
- lytic rib lesion (mnemonic)
- differential diagnosis of epiphyseal overgrowth
- fibrous lesions
- cementifying fibroma
- Knochenläsionen der Diaphyse
- Läsionen der Mandibula
- fibröse Dysplasie am Keilbeinflügel
- Fibröse Dysplasie des proximalen Femurs
- fibrous dysplasia: distal femur
- Fibröse Dysplasie des Femur
- arrested pneumatisation of the skull base
- Mazabraud-Syndrom
- fibröse Dysplasie der Mandibula
- polyostotic craniofacial fibrous dysplasia in infancy
- monostotic fibrous dysplasia of the chest wall
- fibrous dysplasia of the temporal bone
- monostotic fibrous dysplasia with epiphyseal involvement
- monostotic fibrous dysplasia of the mandible
- fibrous dysplasia of the orbital roof
- bony lesions without periostitis or pain (mnemonic)
- Osteom des Orbitadaches
- Knochendysplasie
- Milchglas Knochen
- Polyzystische Lipomembranöse Osteodysplasie mit Sklerotischer Leukoenzephalopathie
- Gnatho-diaphysäre Dysplasie
- Fibröse Dysplasie Szintigraphie
- fibrous dysplasia of the fibula
- fibrous dysplasia diffusion weighted imaging


 Assoziationen und Differentialdiagnosen zu Fibröse Dysplasie:
Assoziationen und Differentialdiagnosen zu Fibröse Dysplasie: